UniVI: a scalable multi-modal variational autoencoder toolkit for seamless integration and analysis of multimodal single-cell data.

Project description
UniVI





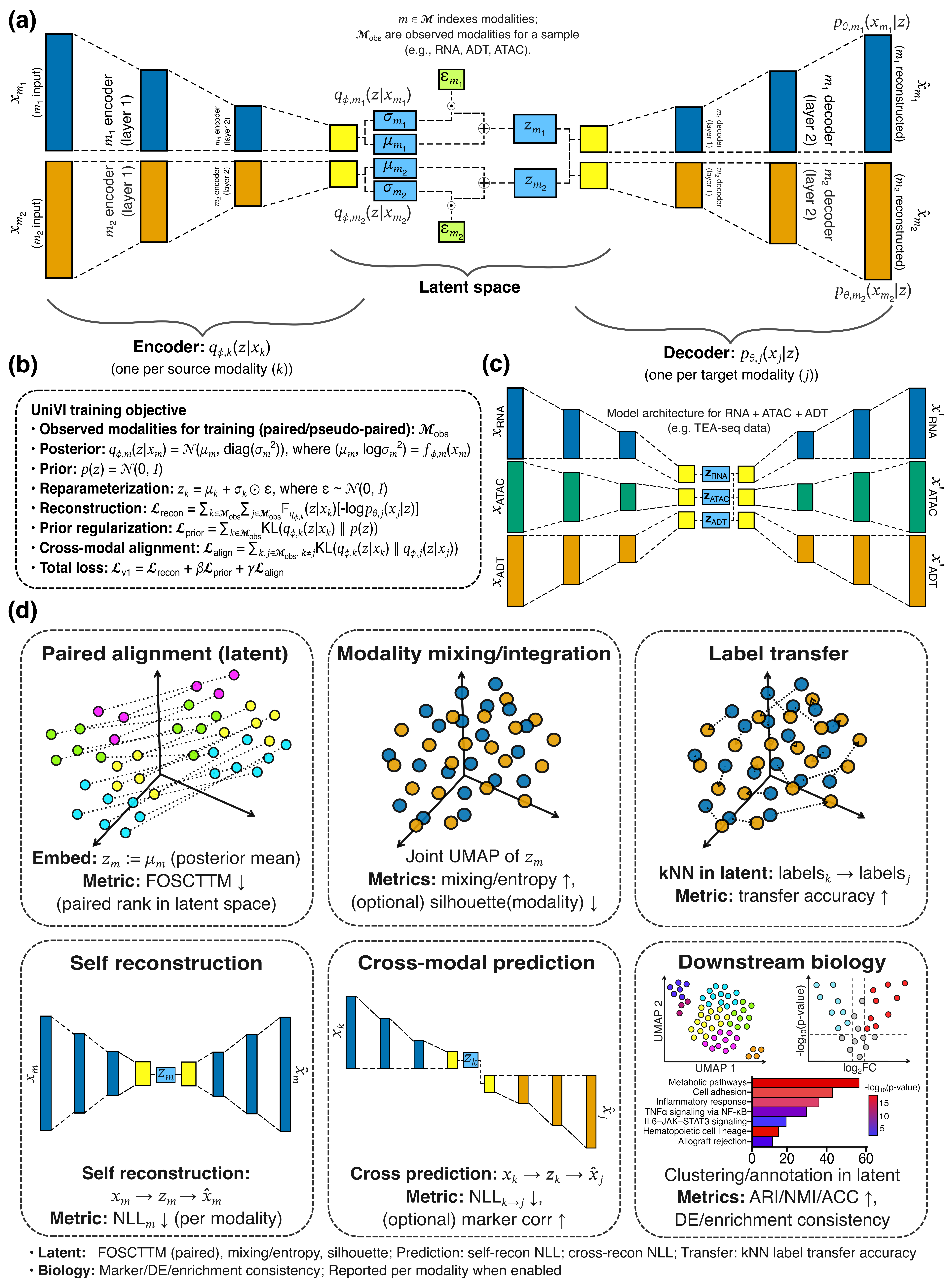
UniVI is a multi-modal variational autoencoder (VAE) toolkit for aligning and integrating single-cell modalities such as RNA, ADT (CITE-seq), ATAC, and coverage-aware / proportion-like assays (e.g., single-cell methylome features).
Common use cases:
- Joint embedding of paired multimodal data (CITE-seq, Multiome, TEA-seq)
- Bridge mapping / projection of unimodal cohorts into a paired latent
- Cross-modal imputation (RNA→ADT, ATAC→RNA, RNA→methylome, …)
- Denoising / reconstruction with likelihood-aware decoders
- Evaluation (FOSCTTM, Recall@k, mixing/entropy, label transfer, clustering)
- Optional supervised heads, MoE gating diagnostics, and transformer encoders
Preprint
If you use UniVI in your work, please cite:
Ashford AJ, Enright T, Somers J, Nikolova O, Demir E.
Unifying multimodal single-cell data with a mixture-of-experts β-variational autoencoder framework.
bioRxiv (2025; updated 2026). doi: 10.1101/2025.02.28.640429
@article{Ashford2025UniVI,
title = {Unifying multimodal single-cell data with a mixture-of-experts β-variational autoencoder framework},
author = {Ashford, A. J. and Enright, T. and Somers, J. and Nikolova, O. and Demir, E.},
journal = {bioRxiv},
date = {2025},
doi = {10.1101/2025.02.28.640429},
url = {https://www.biorxiv.org/content/10.1101/2025.02.28.640429},
note = {Preprint (updated 2026)}
}
Installation
PyPI
pip install univi
UniVI requires PyTorch. If
import torchfails, install PyTorch for your platform/CUDA from PyTorch’s official install instructions.
Conda / mamba
conda install -c conda-forge univi
# or
mamba install -c conda-forge univi
Development install (from source)
git clone https://github.com/Ashford-A/UniVI.git
cd UniVI
conda env create -f envs/univi_env.yml
conda activate univi_env
pip install -e .
Data expectations
UniVI expects per-modality AnnData objects.
- Each modality is an
AnnData - For paired settings, modalities share the same cells (
obs_names, same order) - Raw counts often live in
.layers["counts"] - Model inputs typically live in
.X(or.obsm["X_*"]for ATAC LSI)
Recommended convention:
.layers["counts"]= raw counts / raw signal.X/.obsm["X_*"]= model input space (log1p RNA, CLR ADT, LSI ATAC, methyl fractions, etc.).layers["denoised_*"]/.layers["imputed_*"]= UniVI outputs
Methylome / coverage-aware modalities
Two common patterns:
A) Fraction-valued features (simple path)
If .X contains values in [0, 1] (fraction methylated) and you don’t need coverage-aware likelihoods:
- store fractions in
.X - use
likelihood="beta"
B) Counts + coverage (recommended when available)
If you have both:
- successes (e.g., methylated counts) and
- total_count (coverage / trials)
Use:
likelihood="binomial"orlikelihood="beta_binomial"(often preferred)
In this setup the model input can still be fractions/embeddings in .X, but the reconstruction loss is computed against recon_targets (successes + total_count) supplied by the dataset/collate path.
Quickstart (Python / Jupyter)
Minimal “notebook path”: load paired AnnData → train → encode/evaluate.
import numpy as np
import scanpy as sc
import torch
from torch.utils.data import DataLoader, Subset
from univi import UniVIMultiModalVAE, ModalityConfig, UniVIConfig, TrainingConfig
from univi.data import MultiModalDataset, align_paired_obs_names, collate_multimodal_xy_recon
from univi.trainer import UniVITrainer
1) Load paired AnnData
rna = sc.read_h5ad("path/to/rna_citeseq.h5ad")
adt = sc.read_h5ad("path/to/adt_citeseq.h5ad")
adata_dict = align_paired_obs_names({"rna": rna, "adt": adt})
2) Dataset + dataloaders (MultiModalDataset option)
device = "cuda" if torch.cuda.is_available() else ("mps" if torch.backends.mps.is_available() else "cpu")
dataset = MultiModalDataset(
adata_dict=adata_dict,
X_key="X", # uses .X as model input
device=None, # dataset yields CPU tensors; model moves to GPU
)
n = rna.n_obs
idx = np.arange(n)
rng = np.random.default_rng(0)
rng.shuffle(idx)
split = int(0.8 * n)
train_idx, val_idx = idx[:split], idx[split:]
train_loader = DataLoader(
Subset(dataset, train_idx),
batch_size=256,
shuffle=True,
num_workers=0,
collate_fn=collate_multimodal_xy_recon, # safe even if recon_targets are absent
)
val_loader = DataLoader(
Subset(dataset, val_idx),
batch_size=256,
shuffle=False,
num_workers=0,
collate_fn=collate_multimodal_xy_recon,
)
3) Model config + train
univi_cfg = UniVIConfig(
latent_dim=30,
beta=1.15,
gamma=3.25,
modalities=[
# Likelihood guidance:
# - RNA (normalized/log1p): often "gaussian"
# - RNA (raw counts): "nb" or "zinb"
# - ADT (CLR/scaled): often "gaussian"
# - ATAC (binarized peaks): "bernoulli"
# - ATAC (peak counts): "poisson" (sometimes; often too restrictive if overdispersed)
# - ATAC (LSI / reduced features): often "gaussian" for integration-focused workflows
# - Methylome fractions in (0,1): "beta"
# - Methylome counts+coverage: "binomial" or "beta_binomial" (often preferred)
#
# IMPORTANT for "binomial" / "beta_binomial":
# The reconstruction target must include BOTH successes and total_count
# (passed via `recon_targets` as a keyword argument to the model/training step), e.g.:
# {"successes": m, "total_count": n}
#
# NOTE:
# Manuscript-style "gaussian" decoders on normalized feature spaces often produce the most
# cell-to-cell aligned latent spaces for integration-focused use cases. For some assay types
# (including methylome), a more distribution-matched likelihood may be preferable depending
# on whether your goal is alignment vs calibrated reconstruction/generation.
#
ModalityConfig(
name="rna",
input_dim=rna.n_vars,
encoder_hidden=[1024, 512, 256, 128],
decoder_hidden=[128, 256, 512, 1024],
likelihood="gaussian",
),
ModalityConfig(
name="adt",
input_dim=adt.n_vars,
encoder_hidden=[256, 128, 64],
decoder_hidden=[64, 128, 256],
likelihood="gaussian",
),
],
)
train_cfg = TrainingConfig(
n_epochs=2000,
batch_size=256,
lr=1e-3,
weight_decay=1e-4,
device=device,
early_stopping=True,
best_epoch_warmup=50,
patience=50,
)
model = UniVIMultiModalVAE(
univi_cfg,
loss_mode="v1", # "v1" recommended (used in the manuscript)
v1_recon="avg",
normalize_v1_terms=True,
).to(device)
trainer = UniVITrainer(
model=model,
train_loader=train_loader,
val_loader=val_loader,
train_cfg=train_cfg,
device=device,
)
trainer.fit()
Quickstart: RNA + methylome (beta-binomial with recon_targets)
Assumes:
meth.Xis a convenient model input (fractions or embeddings)meth.layers["meth_successes"]stores methylated countsmeth.layers["meth_total_count"]stores coverage / trials
rna = sc.read_h5ad("path/to/rna.h5ad")
meth = sc.read_h5ad("path/to/meth.h5ad")
adata_dict = align_paired_obs_names({"rna": rna, "meth": meth})
recon_targets_spec = {
"meth": {
"successes_layer": "meth_successes",
"total_count_layer": "meth_total_count",
}
}
dataset = MultiModalDataset(
adata_dict=adata_dict,
X_key="X",
device=None,
recon_targets_spec=recon_targets_spec,
)
train_loader = DataLoader(
dataset,
batch_size=256,
shuffle=True,
num_workers=0,
collate_fn=collate_multimodal_xy_recon,
)
univi_cfg = UniVIConfig(
latent_dim=30,
beta=1.15,
gamma=3.25,
modalities=[
ModalityConfig("rna", rna.n_vars, [1024, 512, 256, 128], [128, 256, 512, 1024], likelihood="gaussian"),
ModalityConfig("meth", meth.n_vars, [512, 256, 128], [128, 256, 512], likelihood="beta_binomial"),
],
)
train_cfg = TrainingConfig(n_epochs=2000, batch_size=256, lr=1e-3, weight_decay=1e-4, device=device)
model = UniVIMultiModalVAE(univi_cfg, loss_mode="v1", v1_recon="avg", normalize_v1_terms=True).to(device)
trainer = UniVITrainer(model=model, train_loader=train_loader, val_loader=None, train_cfg=train_cfg, device=device)
trainer.fit()
When
recon_targetsare present in the batch,UniVITrainerforwards them intomodel(..., recon_targets=...)automatically.
Saving + loading
import torch
from univi import UniVIMultiModalVAE
ckpt = {
"model_state_dict": model.state_dict(),
"model_config": univi_cfg,
"train_cfg": train_cfg,
"history": getattr(trainer, "history", None),
"best_epoch": getattr(trainer, "best_epoch", None),
}
torch.save(ckpt, "./saved_models/univi_model_state.pt")
ckpt = torch.load("./saved_models/univi_model_state.pt", map_location=device)
model = UniVIMultiModalVAE(ckpt["model_config"]).to(device)
model.load_state_dict(ckpt["model_state_dict"])
model.eval()
print("Best epoch:", ckpt.get("best_epoch"))
After training: what you can do with a UniVI model
UniVI models are generative (decoders + likelihoods) and alignment-oriented (shared latent space). After training, you typically use two modules:
univi.evaluation: encoding, denoising, cross-modal prediction (imputation), generation, and metricsunivi.plotting: Scanpy/Matplotlib helpers for UMAPs, legends, confusion matrices, MoE gate plots, and reconstruction-error plots
0) Imports + plotting defaults
import numpy as np
import scipy.sparse as sp
import torch
from univi.evaluation import (
encode_adata,
encode_fused_adata_pair,
cross_modal_predict,
denoise_adata,
denoise_from_multimodal,
evaluate_alignment,
reconstruction_metrics,
# NEW (generation + recon error workflows)
generate_from_latent,
fit_label_latent_gaussians,
sample_latent_by_label,
evaluate_cross_reconstruction,
)
from univi.plotting import (
set_style,
umap,
umap_by_modality,
compare_raw_vs_denoised_umap_features,
plot_confusion_matrix,
write_gates_to_obs,
plot_moe_gate_summary,
# NEW (reconstruction error plots)
plot_reconstruction_error_summary,
plot_featurewise_reconstruction_scatter,
)
set_style(font_scale=1.2, dpi=150)
device = "cuda" if torch.cuda.is_available() else ("mps" if torch.backends.mps.is_available() else "cpu")
Helper for sparse matrices:
def to_dense(X):
return X.toarray() if sp.issparse(X) else np.asarray(X)
1) Encode a modality into latent space (.obsm["X_univi"])
Use this when you have one observed modality at a time (RNA-only, ADT-only, ATAC-only, methylome-only, etc.):
Z_rna = encode_adata(
model,
adata=rna,
modality="rna",
device=device,
layer=None, # uses adata.X by default
X_key="X",
batch_size=1024,
latent="moe_mean", # {"moe_mean","moe_sample","modality_mean","modality_sample"}
random_state=0,
)
rna.obsm["X_univi"] = Z_rna
Then plot:
umap(
rna,
obsm_key="X_univi",
color=["celltype.l2", "batch"],
legend="outside",
legend_subset_topk=25,
savepath="umap_rna_univi.png",
show=False,
)
2) Encode a fused multimodal latent (true paired/multi-observed cells)
When you have multiple observed modalities for the same cells, you can encode the fused posterior (and optionally MoE router gates/logits):
fused = encode_fused_adata_pair(
model,
adata_by_mod={"rna": rna, "adt": adt}, # same obs_names, same order
device=device,
batch_size=1024,
use_mean=True,
return_gates=True,
return_gate_logits=True,
write_to_adatas=True, # writes obsm + gate columns
fused_obsm_key="X_univi_fused",
gate_prefix="gate",
)
# fused["Z_fused"] -> (n_cells, latent_dim)
# fused["gates"] -> (n_cells, n_modalities) or None (if fused transformer posterior is used)
Plot fused:
umap(
rna,
obsm_key="X_univi_fused",
color=["celltype.l2", "batch"],
legend="outside",
savepath="umap_fused.png",
show=False,
)
Plot fused both modalities by modality and celltype:
umap_by_modality(
{"rna": rna, "adt": adt},
obsm_key="X_univi_fused",
color=["univi_modality", "celltype.l2"],
legend="outside",
size=8,
savepath="umap_fused_both_modalities.png",
show=False,
)
3) Cross-modal prediction (imputation): encode source → decode target
Example: RNA → ADT (same pattern applies to RNA→methylome, methylome→RNA, etc.). UniVI will automatically handle decoder output types internally (e.g. Gaussian returns tensor; NB returns {"mu","log_theta"}; ZINB returns {"mu","log_theta","logit_pi"}; Poisson returns {"rate","log_rate"}; Beta/Beta-Binomial return parameter dicts) and return an appropriate mean-like prediction for downstream evaluation/plotting.
adt_hat_from_rna = cross_modal_predict(
model,
adata_src=rna,
src_mod="rna",
tgt_mod="adt",
device=device,
layer=None,
X_key="X",
batch_size=512,
use_moe=True,
)
adt.layers["imputed_from_rna"] = adt_hat_from_rna
4) Denoising (self-reconstruction or true fused denoising)
Option A — self-denoise a single modality (same as “reconstruct”)
denoise_adata(
model,
adata=rna,
modality="rna",
device=device,
out_layer="denoised_self",
overwrite_X=False,
batch_size=512,
)
Option B — true multimodal denoising via fused latent
denoise_adata(
model,
adata=rna, # output written here
modality="rna",
device=device,
out_layer="denoised_fused",
overwrite_X=False,
batch_size=512,
adata_by_mod={"rna": rna, "adt": adt},
layer_by_mod={"rna": None, "adt": None}, # None -> use .X
X_key_by_mod={"rna": "X", "adt": "X"},
use_mean=True,
)
Compare raw vs denoised marker overlays:
compare_raw_vs_denoised_umap_features(
rna,
obsm_key="X_univi",
features=["MS4A1", "CD3D", "NKG7"],
raw_layer=None,
denoised_layer="denoised_fused",
savepath="umap_raw_vs_denoised.png",
show=False,
)
5) Quantify reconstruction / imputation error vs ground truth
You can compute featurewise + summary errors between:
- cross-reconstructed (RNA→ADT, ATAC→RNA, methylome→RNA, …)
- denoised outputs (self or fused)
- and the true observed data
A) Basic metrics on two matrices
true = to_dense(adt.X)
pred = adt.layers["imputed_from_rna"]
m = reconstruction_metrics(true, pred)
print("MSE mean:", m["mse_mean"])
print("Pearson mean:", m["pearson_mean"])
B) One-call evaluation for cross-reconstruction / denoising
This will:
- generate predictions via UniVI (handling decoder output types correctly),
- align to the requested truth matrix (layer/X_key), and
- return metrics + optional per-feature vectors.
rep = evaluate_cross_reconstruction(
model,
adata_src=rna,
adata_tgt=adt,
src_mod="rna",
tgt_mod="adt",
device=device,
src_layer=None,
tgt_layer=None,
batch_size=512,
# optionally restrict to a feature subset (e.g., top markers)
feature_names=None,
)
print(rep["summary"]) # mse_mean/median, pearson_mean/median, etc.
Plot reconstruction-error summaries:
plot_reconstruction_error_summary(
rep,
title="RNA → ADT imputation error",
savepath="recon_error_summary.png",
show=False,
)
And featurewise scatter (true vs predicted) for selected features:
plot_featurewise_reconstruction_scatter(
rep,
features=["CD3", "CD4", "MS4A1"],
savepath="recon_scatter_selected_features.png",
show=False,
)
6) Alignment evaluation (FOSCTTM, Recall@k, mixing/entropy, label transfer, gates)
metrics = evaluate_alignment(
Z1=rna.obsm["X_univi"],
Z2=adt.obsm["X_univi"],
metric="euclidean",
recall_ks=(1, 5, 10),
k_mixing=20,
k_entropy=30,
labels_source=rna.obs["celltype.l2"].to_numpy(),
labels_target=adt.obs["celltype.l2"].to_numpy(),
compute_bidirectional_transfer=True,
k_transfer=15,
json_safe=True,
)
Confusion matrix:
plot_confusion_matrix(
np.asarray(metrics["label_transfer_cm"]),
labels=np.asarray(metrics["label_transfer_label_order"]),
title="Label transfer (RNA → ADT)",
normalize="true",
savepath="label_transfer_confusion.png",
show=False,
)
7) Generate new data from latent space (sampling / “in silico cells”)
UniVI decoders define a likelihood per modality (Gaussian, NB, ZINB, Poisson, Bernoulli, Beta, Binomial/Beta-Binomial, etc.). Generation is done as:
- pick latent samples
z ~ p(z)(or a conditional latent distribution) - decode with the modality decoder(s)
- return mean-like reconstructions or (optionally) sample from the likelihood
A) Unconditional generation (standard normal prior)
Xgen = generate_from_latent(
model,
n=5000,
target_mod="rna",
device=device,
z_source="prior", # "prior" or provide z directly
return_mean=True, # mean-like output
sample_likelihood=False, # if True: sample from likelihood when supported
)
# Xgen shape: (5000, n_genes)
B) Cell-type–conditioned generation via empirical latent neighborhoods
This is the “no classifier head needed” option:
- encode a reference cohort
- pick cells with a given label
- sample around their latent distribution (Gaussian fit, or jitter)
Z = rna.obsm["X_univi"]
labels = rna.obs["celltype.l2"].to_numpy()
# Fit a per-label Gaussian in latent space
label_gauss = fit_label_latent_gaussians(Z, labels)
# Sample latent points for a chosen label
z_B = sample_latent_by_label(label_gauss, label="B cell", n=2000, random_state=0)
# Decode to RNA space
X_B = generate_from_latent(
model,
z=z_B,
target_mod="rna",
device=device,
return_mean=True,
)
C) Cluster-aware generation (no annotations required)
If you don’t have labels, you can cluster Z (e.g., k-means), fit cluster Gaussians, then sample by cluster id.
D) Head-guided generation (optional, when a classifier head exists)
If you trained a classification head, you can optionally bias latent selection toward a desired label by filtering or optimizing candidate z’s (implementation depends on your head setup). UniVI supports this workflow when the head is present, but the label-agnostic Gaussian/cluster methods work everywhere.
8) MoE gating diagnostics (precision contributions + optional learnable router)
UniVI can report per-cell modality contribution weights for the analytic fusion path (MoE/PoE-style).
There are two related notions of “who contributed how much” to the fused latent:
- Precision-only (always available): derived from each modality’s posterior uncertainty in latent space.
- Router × precision (optional): if your trained model exposes router logits, UniVI can combine router probabilities with precision to produce contribution weights.
Note: This section applies to analytic fusion (Gaussian experts in latent space). If you use a fused transformer posterior, there may be no analytic precision/router attribution and gates can be unavailable or not meaningful.
A) Compute per-cell contribution weights (recommended)
from univi.evaluation import to_dense, encode_moe_gates_from_tensors
from univi.plotting import write_gates_to_obs, plot_moe_gate_summary
gate = encode_moe_gates_from_tensors(
model,
x_dict={"rna": to_dense(rna.X), "adt": to_dense(adt.X)},
device=device,
batch_size=1024,
modality_order=["rna", "adt"],
kind="router_x_precision", # falls back to "effective_precision" if router logits are unavailable
return_logits=True,
)
W = gate["weights"] # (n_cells, n_modalities), rows sum to 1
mods = gate["modality_order"] # e.g. ["rna", "adt"]
print("Requested kind:", gate.get("requested_kind"))
print("Effective kind:", gate.get("kind"))
print("Per-modality mean:", gate.get("per_modality_mean"))
print("Has logits:", gate.get("logits") is not None)
If you want precision-only weights (no router influence), set kind="effective_precision".
B) Write weights to .obs (for plotting / grouping)
write_gates_to_obs(
rna,
gates=W,
modality_names=mods,
gate_prefix="moe_gate", # creates obs cols: moe_gate_{mod}
gate_logits=gate.get("logits"), # optional; may be None
)
C) Plot contribution usage (overall + grouped)
plot_moe_gate_summary(
rna,
gate_prefix="moe_gate",
groupby="celltype.l3", # or "celltype.l2", "batch", etc.
agg="mean",
savepath="moe_gates_by_celltype.png",
show=False,
)
D) Optional: log gates alongside alignment metrics
evaluate_alignment(...) evaluates geometric alignment (FOSCTTM, Recall@k, mixing/entropy, label transfer).
If you want to save gate summaries alongside those metrics, just merge dictionaries:
from univi.evaluation import evaluate_alignment
metrics = evaluate_alignment(
Z1=rna.obsm["X_univi"],
Z2=adt.obsm["X_univi"],
labels_source=rna.obs["celltype.l3"].to_numpy(),
labels_target=adt.obs["celltype.l3"].to_numpy(),
json_safe=True,
)
metrics["moe_gates"] = {
"kind": gate.get("kind"),
"requested_kind": gate.get("requested_kind"),
"modality_order": mods,
"per_modality_mean": gate.get("per_modality_mean"),
# (optional) store full matrices; omit if you want small JSON
# "weights": W,
# "logits": gate.get("logits"),
}
Advanced topics
Training objectives (v1 vs v2/lite)
- v1 (“paper”): per-modality posteriors + reconstruction scheme (cross/self/avg) + posterior alignment
- v2/lite: fused posterior (MoE/PoE by default; optional fused transformer) + per-modality recon + β·KL + γ·alignment
Choose via loss_mode at construction time (Python) or config JSON (scripts).
Decoder output types (what UniVI handles for you)
Decoders can return either:
- a tensor (e.g. Gaussian)
- or a dict (e.g. NB/ZINB/Poisson/Beta/Beta-Binomial parameter dicts)
UniVI evaluation utilities unwrap these and return mean-like matrices for plotting/evaluation.
Advanced topics
Training objectives (v1 vs v2/lite)
- v1 (“paper”): per-modality posteriors + reconstruction scheme (cross/self/avg) + posterior alignment across modalities
- v2/lite: fused posterior (MoE/PoE-style by default; optional fused transformer) + per-modality recon + β·KL + γ·alignment (L2 on latent means)
Choose via loss_mode at construction time (Python) or config JSON (scripts).
Decoder output types (what UniVI handles for you)
Decoders can return either:
- a tensor (e.g. Gaussian)
- or a dict (e.g. NB/ZINB/Poisson/Beta/Beta-Binomial parameter dicts)
UniVI evaluation utilities unwrap these and return mean-like matrices for plotting/evaluation.
Advanced model features
This section covers the “advanced” knobs in univi/models/univi.py and when to use them. Everything below is optional: you can train and evaluate UniVI without touching any of it.
1) Fused multimodal transformer posterior (optional)
What it is:
A single fused encoder that tokenizes each observed modality, concatenates tokens, runs a multimodal transformer, and outputs a fused posterior (mu_fused, logvar_fused).
Why you’d use it:
- You want the posterior to be learned jointly across modalities (rather than fused analytically via PoE/MoE precision fusion).
- You want token-level interpretability hooks (e.g., ATAC top-k peak indices; optional attention maps if enabled in the encoder stack).
- You want a learnable “cross-modality mixing” mechanism beyond precision fusion.
How to enable (config):
-
Set
cfg.fused_encoder_type = "multimodal_transformer". -
Optionally set:
cfg.fused_modalities = ["rna","adt","atac"](defaults to all)cfg.fused_require_all_modalities = True(default): only use fused posterior when all required modalities are present; otherwise falls back tomixture_of_experts().
Key API points:
- Training: the model will automatically decide whether to use fused encoder or fallback based on presence and
fused_require_all_modalities. - Encoding: use
model.encode_fused(...)to get the fused latent and optionally gates from fallback fusion.
mu, logvar, z = model.encode_fused(
{"rna": X_rna, "adt": X_adt, "atac": X_atac},
use_mean=True,
)
2) Attention bias for transformer encoders (distance bias for ATAC, optional)
What it is: A safe, optional attention bias that can encourage local genomic context for tokenized ATAC (or any modality tokenizer that supports it). It’s a no-op unless:
- the encoder is transformer-based and
- its tokenizer exposes
build_distance_attn_bias()and - you pass
attn_bias_cfg.
Why you’d use it:
- ATAC token sets are sparse and positional: distance-aware attention can help the transformer focus on local regulatory structure.
How to use (forward / encode / predict):
Pass attn_bias_cfg into forward(...), encode_fused(...), or predict_heads(...).
attn_bias_cfg = {
"atac": {"type": "distance", "lengthscale_bp": 50_000, "same_chrom_only": True}
}
out = model(x_dict=x_dict, epoch=ep, attn_bias_cfg=attn_bias_cfg)
mu, logvar, z = model.encode_fused(x_dict, attn_bias_cfg=attn_bias_cfg)
pred = model.predict_heads(x_dict, attn_bias_cfg=attn_bias_cfg)
Notes:
- For the fused multimodal transformer posterior, UniVI applies distance bias within the ATAC token block and leaves cross-modality blocks neutral (0), so it won’t artificially “force” cross-modality locality.
3) Learnable MoE gating for fusion (optional)
What it is: A learnable gate that produces per-cell modality weights and uses them to scale per-modality precisions before PoE-style fusion. This is off by default; without it, fusion is pure precision fusion.
Why you’d use it:
- Modalities have variable quality per cell (e.g., low ADT counts, sparse ATAC, stressed RNA, low methylome coverage).
- You want a data-driven “trust score” per modality per cell.
- You want interpretable per-cell reliance weights (gate weights) to diagnose integration behavior.
How to enable (config):
-
cfg.use_moe_gating = True -
Optional:
cfg.moe_gating_type = "per_modality"(default) or"shared"cfg.moe_gating_hidden = [..],cfg.moe_gating_dropout,cfg.moe_gating_batchnorm,cfg.moe_gating_activationcfg.moe_gate_epsto avoid exact zeros in gated precisions
How to retrieve gates:
Use encode_fused(..., return_gates=True) (works when not using fused transformer posterior; if fused posterior is used, gates are None).
mu, logvar, z, gates, gate_logits = model.encode_fused(
x_dict,
use_mean=True,
return_gates=True,
return_gate_logits=True,
)
# gates: (n_cells, n_modalities) in the model's modality order
Tip: Gate weights are useful for plots like “ADT reliance by celltype” or identifying low-quality subsets.
4) Multi-head supervised decoders (classification + adversarial heads)
UniVI supports two supervised head systems:
A) Legacy single label head (kept for backwards compatibility)
What it is:
A single categorical head via label_decoder controlled by init args:
n_label_classes,label_loss_weight,label_ignore_index,classify_from_mu,label_head_name
When to use it: If you already rely on the legacy label head in notebooks/scripts and want a stable API.
Label names helpers:
model.set_label_names(["B", "T", "NK", ...])
B) New cfg.class_heads multi-head system (recommended for new work)
What it is:
Any number of heads defined via ClassHeadConfig. Heads can be:
- categorical: softmax + cross-entropy
- binary: single logit + BCEWithLogitsLoss (optionally with
pos_weight)
Heads can also be adversarial: they apply a gradient reversal layer (GRL) to encourage invariance (domain confusion).
Why you’d use it:
- Predict multiple labels simultaneously (celltype, batch, donor, tissue, QC flags, etc.).
- Add domain-adversarial training (e.g., suppress batch/donor information).
- Semi-supervised setups where only some labels exist per head.
How labels are passed at training time:
y should be a dict keyed by head name:
y = {
"celltype": celltype_ids, # categorical (shape [B] or one-hot [B,C])
"batch": batch_ids, # adversarial categorical, for batch-invariant latents
"is_doublet": doublet_01, # binary head (0/1, ignore_index supported)
}
out = model(x_dict=x_dict, epoch=ep, y=y)
How to predict heads after training:
Use predict_heads(...) to run encoding + head prediction in one call.
pred = model.predict_heads(x_dict, return_probs=True)
# pred[head] returns probabilities (softmax for categorical, sigmoid for binary)
Head label name helpers (categorical):
model.set_head_label_names("celltype", ["B", "T", "NK", ...])
Inspect head configuration (useful for logging):
meta = model.get_classification_meta()
5) Label expert injection into the fused posterior (semi-supervised “label as expert”)
What it is: Optionally treats labels as an additional expert by encoding the label into a Gaussian posterior and fusing it with the base fused posterior. Controlled by:
use_label_encoder=Trueandn_label_classes>0label_encoder_warmup(epoch threshold before injection starts)label_moe_weight(how strong labels influence fusion)unlabeled_logvar(large => labels contribute little when missing)
Why you’d use it:
- Semi-supervised alignment: labels can stabilize the latent when paired signals are weak.
- Controlled injection after warmup to avoid early collapse.
How to use in encoding:
encode_fused(..., inject_label_expert=True, y=...)
mu, logvar, z = model.encode_fused(
x_dict,
epoch=ep,
y={"label": y_ids}, # or just pass y_ids if using legacy path
inject_label_expert=True,
)
6) Recon scaling across modalities (important when dims differ a lot)
What it is: Per-modality reconstruction losses are typically summed across features; large modalities (RNA) can dominate gradients. UniVI supports:
recon_normalize_by_dim+recon_dim_power(divide byD**power)- per-modality
ModalityConfig.recon_weight
Defaults:
- v1-style losses: normalize is off by default,
power=0.5 - v2/lite: normalize is on by default,
power=1.0
Why you’d use it:
- Stabilize training when RNA has 2k–20k dims but ADT has 30–200 dims and ATAC-LSI has ~50–500 dims (and methylome features may vary widely too).
- Tune modality balance without hand-waving.
How to tune:
- For “equal per-cell contribution” across modalities:
recon_normalize_by_dim=Trueandrecon_dim_power=1.0 - If you want a softer correction:
power=0.5 - Or set
recon_weightper modality.
7) Convenience APIs
encode_fused(...)
Purpose: Encode any subset of modalities into a fused posterior, with optional gate outputs.
mu, logvar, z = model.encode_fused(
x_dict,
epoch=0,
use_mean=True, # True: return mu; False: sample
inject_label_expert=True,
attn_bias_cfg=None,
)
# Optional: get fusion gates (only when fused transformer posterior is NOT used)
mu, logvar, z, gates, gate_logits = model.encode_fused(
x_dict,
return_gates=True,
return_gate_logits=True,
)
predict_heads(...)
Purpose: Encode fused latent, then emit probabilities/logits for the legacy head + all multi-head configs.
pred = model.predict_heads(x_dict, return_probs=True)
# pred[head] -> probs (softmax/sigmoid)
Repository structure
UniVI/
├── README.md # Project overview, installation, quickstart
├── LICENSE # MIT license text file
├── pyproject.toml # Python packaging config (pip / PyPI)
├── assets/ # Static assets used by README/docs
│ └── figures/ # Schematic figure(s) for repository front page
├── conda.recipe/ # Conda build recipe (for conda-build)
│ └── meta.yaml
├── envs/ # Example conda environments
│ ├── UniVI_working_environment.yml
│ ├── UniVI_working_environment_v2_full.yml
│ ├── UniVI_working_environment_v2_minimal.yml
│ └── univi_env.yml # Recommended env (CUDA-friendly)
├── data/ # Small example data notes (datasets are typically external)
│ └── README.md # Notes on data sources / formats
├── notebooks/ # End-to-end Jupyter Notebook analyses and examples
│ ├── GR_manuscript_reproducibility/ # Reproduce figures from revised manuscript (in progress for Genome Research; bioRxiv manuscript v2)
│ │ ├── UniVI_manuscript_GR-Figure__2__CITE_paired.ipynb
│ │ ├── UniVI_manuscript_GR-Figure__3__CITE_paired_biological_latent.ipynb
│ │ ├── UniVI_manuscript_GR-Figure__4__Multiome_paired.ipynb
│ │ ├── UniVI_manuscript_GR-Figure__5__Multiome_bridge_mapping_and_fine-tuning.ipynb
│ │ ├── UniVI_manuscript_GR-Figure__6__TEA-seq_tri-modal.ipynb
│ │ ├── UniVI_manuscript_GR-Figure__7__AML_bridge_mapping_and_fine-tuning.ipynb
│ │ ├── UniVI_manuscript_GR-Figure__8__benchmarking_against_pytorch_tools.ipynb
│ │ ├── UniVI_manuscript_GR-Figure__8__benchmarking_against_R_tools.ipynb
│ │ ├── UniVI_manuscript_GR-Figure__8__benchmarking_merging_and_plotting_runs.ipynb
│ │ ├── UniVI_manuscript_GR-Figure__9__paired_data_ablation_and_computational_scaling_performance.ipynb
│ │ ├── UniVI_manuscript_GR-Figure__9__paired_data_ablation_and_computational_scaling_performance_compile_plots_from_results_df.ipynb
│ │ ├── UniVI_manuscript_GR-Figure_10__cell_population_ablation_MoE.ipynb
│ │ ├── UniVI_manuscript_GR-Figure_10__cell_population_ablation_MoE_compile_plots_from_results_df.ipynb
│ │ ├── UniVI_manuscript_GR-Supple_____grid-sweep.ipynb
│ │ └── UniVI_manuscript_GR-Supple_____grid-sweep_compile_plots_from_results_df.ipynb
│ └── UniVI_additional_examples/ # Additional examples of UniVI workflow functionality
│ └── Multiome_NB-RNA-counts_Poisson_or_Bernoulli-ATAC_peak-counts_Peak_perturbation_to_RNA_expression_cross-generation_experiment.ipynb
├── parameter_files/ # JSON configs for model + training + data selectors
│ ├── defaults_*.json # Default configs (per experiment)
│ └── params_*.json # Example “named” configs (RNA, ADT, ATAC, methylome, etc.)
├── scripts/ # Reproducible entry points (revision-friendly)
│ ├── train_univi.py # Train UniVI from a parameter JSON
│ ├── evaluate_univi.py # Evaluate trained models (FOSCTTM, label transfer, etc.)
│ ├── benchmark_univi_citeseq.py # CITE-seq-specific benchmarking script
│ ├── run_multiome_hparam_search.py
│ ├── run_frequency_robustness.py # Composition/frequency mismatch robustness
│ ├── run_do_not_integrate_detection.py # “Do-not-integrate” unmatched population demo
│ ├── run_benchmarks.py # Unified wrapper (includes optional Harmony baseline)
│ └── revision_reproduce_all.sh # One-click: reproduces figures + supplemental tables
└── univi/ # UniVI Python package (importable as `import univi`)
├── __init__.py # Package exports and __version__
├── __main__.py # Enables: `python -m univi ...`
├── cli.py # Minimal CLI (e.g., export-s1, encode)
├── pipeline.py # Config-driven model+data loading; latent encoding helpers
├── diagnostics.py # Exports Supplemental_Table_S1.xlsx (env + hparams + dataset stats)
├── config.py # Config dataclasses (UniVIConfig, ModalityConfig, TrainingConfig)
├── data.py # Dataset wrappers + matrix selectors (layer/X_key, obsm support)
├── evaluation.py # Metrics (FOSCTTM, mixing, label transfer, feature recovery)
├── matching.py # Modality matching / alignment helpers
├── objectives.py # Losses (ELBO variants, KL/alignment annealing, etc.)
├── plotting.py # Plotting helpers + consistent style defaults
├── trainer.py # UniVITrainer: training loop, logging, checkpointing
├── interpretability.py # Helper scripts for transformer token weight interpretability
├── figures/ # Package-internal figure assets (placeholder)
│ └── .gitkeep
├── models/ # VAE architectures + building blocks
│ ├── __init__.py
│ ├── mlp.py # Shared MLP building blocks
│ ├── encoders.py # Modality encoders (MLP + transformer + fused transformer)
│ ├── decoders.py # Likelihood-specific decoders (NB, ZINB, Gaussian, Beta, Binomial/Beta-Binomial, etc.)
│ ├── transformer.py # Transformer blocks + encoder (+ optional attn bias support)
│ ├── tokenizer.py # Tokenization configs/helpers (top-k / patch)
│ └── univi.py # Core UniVI multi-modal VAE
├── hyperparam_optimization/ # Hyperparameter search scripts
│ ├── __init__.py
│ ├── common.py
│ ├── run_adt_hparam_search.py
│ ├── run_atac_hparam_search.py
│ ├── run_citeseq_hparam_search.py
│ ├── run_multiome_hparam_search.py
│ ├── run_rna_hparam_search.py
│ └── run_teaseq_hparam_search.py
└── utils/ # General utilities
├── __init__.py
├── io.py # I/O helpers (AnnData, configs, checkpoints)
├── logging.py # Logging configuration / progress reporting
├── seed.py # Reproducibility helpers (seeding RNGs)
├── stats.py # Small statistical helpers / transforms
└── torch_utils.py # PyTorch utilities (device, tensor helpers)
License
MIT License — see LICENSE.
Contact, questions, and bug reports
-
Questions / comments: open a GitHub Issue with the
questionlabel (or use Discussions) -
Bug reports: include:
- UniVI version:
python -c "import univi; print(univi.__version__)" - minimal notebook/code snippet
- stack trace + OS/CUDA/PyTorch versions
- UniVI version:
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file univi-0.4.7.tar.gz.
File metadata
- Download URL: univi-0.4.7.tar.gz
- Upload date:
- Size: 131.5 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.10.19
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
41f8ac6f79b1a672c95cb08611c8be8d0df65cdc8b9208399fa98e0bec8dc10c
|
|
| MD5 |
0e5dbfba271c555a878058d3553307ab
|
|
| BLAKE2b-256 |
6c05308250269a0f82a0eea687d01c5083874416494d309ac32f07019de0c48a
|
File details
Details for the file univi-0.4.7-py3-none-any.whl.
File metadata
- Download URL: univi-0.4.7-py3-none-any.whl
- Upload date:
- Size: 121.6 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.10.19
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
cb933ebdd74e5fb5cda439a56069911f45cc6edcb24aa05bcdf4e0ded07d1503
|
|
| MD5 |
6d4b6e917ae7540cc3284ea34d72e17a
|
|
| BLAKE2b-256 |
12b96294782c16415e76831fd8ce407a9749583eec1c0ac0a70d7647b979badd
|







