automatic context-specific network inference

Project description
ACSNI
Automatic context-specific network inference
Determining tissue- and disease-specific circuit of biological pathways remains a fundamental goal of molecular biology. Many components of these biological pathways still remain unknown, hindering the full and accurate characterisation of biological processes of interest. ACSNI leverages artificial intelligence for the reconstruction of a biological pathway, aids the discovery of pathway components and classification of the crosstalk between pathways in specific tissues.
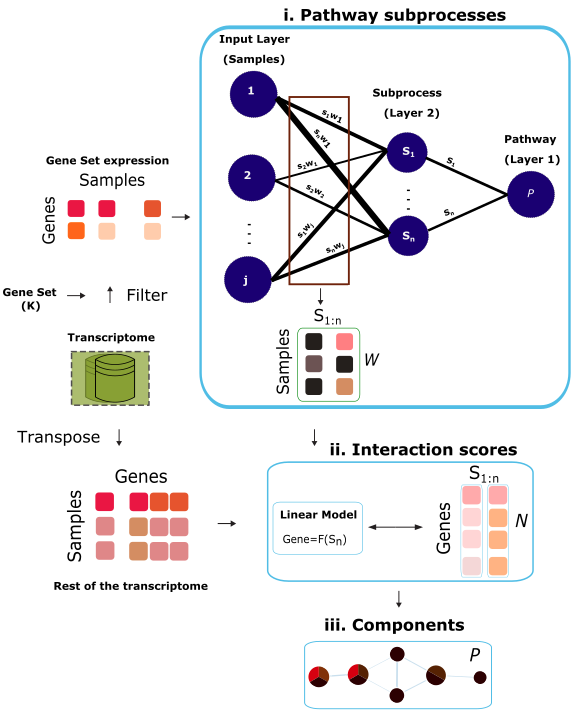
This tool is built in python3.8 with tensorflow backend and keras functional API.
Installation and running the tool
The best way to get ACSNI along with all the dependencies is to install the release from python package installer (pip)
pip install ACSNI
This will add four command line scripts:
| Script | Context | Usage |
|---|---|---|
| ACSNI-run | Gene set analysis | ACSNI-run -h |
| ACSNI-derive | Single gene analysis | ACSNI-derive -h |
| ACSNI-get | Link pathway trait | ACSNI-get -h |
| ACSNI-split | Split expression data | ACSNI-split -h |
Utility functions can be imported using conventional python system like from ACSNI.dbs import ACSNIResults
Input ACSNI-run
Expression Matrix - The expression file (.csv), specified by -i, where columns are samples and rows are genes.
The expression values should be normalised (eg. TPM, CPM, RSEM). Make sure the column name of the 1st column is "gene".
| gene | Sample1 | Sample2 | Sample3 |
|---|---|---|---|
| Foxp1 | 123.2 | 274.1 | 852.6 |
| PD1 | 324.2 | 494.1 | 452.6 |
| CD8 | 523.6 | 624.1 | 252.6 |
This input should not be transformed in any way (e.g. log, z-scale)
Gene set matrix - The prior matrix (.csv) file, specified by -t, where rows are genes and column is a binary
pathway membership. Where "1" means that a gene is in the pathway and "0" means that the gene is not know a priori.
The standard prior looks like below. Make sure the column name of the 1st column is "gene".
| gene | Pathway |
|---|---|
| Foxp1 | 0 |
| PD1 | 0 |
| CD8 | 1 |
You can also supply gene IDs instead of gene symbols.
The tool can handle multiple pathway columns in the -t file as below.
| gene | Pathway1 | Pathway2 | Pathway3 |
|---|---|---|---|
| Foxp1 | 0 | 0 | 0 |
| PD1 | 0 | 1 | 0 |
| CD8 | 1 | 0 | 1 |
Note: Each pathway above is analysed independently, and the outputs have no in-built relationship. The tool is designed to get a granular view of a single pathway at a time.
Output ACSNI-run
Database (.ptl)
| Content | Information |
|---|---|
| co | Pathway Code |
| w | Subprocess space |
| n | Interaction scores |
| p | Score classification |
| d | Interaction direction |
| run_info | Run parameters |
| methods | Extractor functions |
Predicted Network (.csv)
| Content | Meaning |
|---|---|
| name | Gene |
| sub | Subprocess |
| direction | Direction of interactions with subprocess |
Null (.csv) {Shuffled expression matrix}
Input ACSNI-derive
Expression Matrix - See ``-i``` description above.
Note - We recommend removing any un-desirable genes (eg. MT, RPL) from the expression matrix prior to running ACSNI-derive as they usually interfere during initial prior matrix generation steps. For TCR/BCR genes, counts of alpha, beta and gamma chains can be combined into a single count.
Biotype file (Optional) - The biotype file (.csv) specified by -f, given if the generation of gene set should be
based on a particular biotype specified by -b.
| gene | biotype |
|---|---|
| Foxp1 | protein_coding |
| PD1 | protein_coding |
| MALAT1 | lncRNA |
| SNHG12 | lncRNA |
| RNU1-114P | snRNA |
Correlation file (Optional) - The correlation file (.csv) specified by -u, given if the user wishes to replace
"some" specific genes with other genes to be used as a prior for the first iteration of ACSNI-run (internally).
| gene | cor |
|---|---|
| Foxp1 | 0.9 |
| PD1 | 0.89 |
| MALAT1 | 0.85 |
| SNHG12 | 0.80 |
| RNU1-114P | 0.72 |
Output ACSNI-derive
Database (.ptl)
| Content | Information |
|---|---|
| co | Pathway Code |
| n | Interaction scores |
| d | Interaction direction |
| ac | Correlation and T test results |
| fd | Unfiltered prediction data |
| run_info | Run parameters |
| methods | Extractor functions |
Predicted (.csv)
| Content | Meaning |
|---|---|
| name | Gene |
| predict | Classification of genes |
Null (.csv) {Shuffled expression matrix}
Input ACSNI-get
ACSNI database - Output of ACSNI-run (.ptl) specified by -r.
Target phenotype - Biological phenotype file (.csv) to link ACSNI subprocesses, specified by -v.
The sample IDs should match the IDs in the -i analysed by ACSNI-run.
Variable type - The type of phenotype i.e "numeric" or "character", specified by -c.
Outputs the strength of the associations across the subprocesses (.csv).
Input ACSNI-split
Expression Matrix - See ``-i``` description above.
Number of splits - The number of independent cohorts to generate from `-i```.
Outputs the data splits in the current working directory.
Extras
R functions to reproduce the downstream analyses reported in the paper are inside the folder "R".
Example runs are inside the folder "sh".
Tutorial
An extensive tutorial on how to use ACSNI commands can be found inside the Tutorial folder.
To clone the source repository
git clone https://github.com/caanene1/ACSNI
Citation
ACSNI: An unsupervised machine-learning tool for prediction of tissue-specific pathway components using gene expression profiles Chinedu Anthony Anene, Faraz Khan, Findlay Bewicke-Copley, Eleni Maniati and Jun Wang
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file ACSNI-1.0.6.tar.gz.
File metadata
- Download URL: ACSNI-1.0.6.tar.gz
- Upload date:
- Size: 19.3 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.4.1 importlib_metadata/3.7.3 pkginfo/1.7.0 requests/2.25.0 requests-toolbelt/0.9.1 tqdm/4.56.0 CPython/3.8.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
9d8dec7d2c79d1999bd7d0c901e74244c26138da597d941fa7ba50aa0ddf9a68
|
|
| MD5 |
5a9253cacccb003eb282829fe49e16ce
|
|
| BLAKE2b-256 |
74a02105902b302f3845e34989475fd5fd66dcad2a3852320cce1d8db9f0cfd1
|
File details
Details for the file ACSNI-1.0.6-py3-none-any.whl.
File metadata
- Download URL: ACSNI-1.0.6-py3-none-any.whl
- Upload date:
- Size: 23.1 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.4.1 importlib_metadata/3.7.3 pkginfo/1.7.0 requests/2.25.0 requests-toolbelt/0.9.1 tqdm/4.56.0 CPython/3.8.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
3498e2510cab07590f218b4df54c58dce1149f1dc46d30661fdd7c4f18af0109
|
|
| MD5 |
b2716f5a97718b42a92cae36f86c71f1
|
|
| BLAKE2b-256 |
98ecfc29fe99de92955a53ca19d7c12f781f140f8f2a0e835b8042758deb8fd3
|







