BiGAnts - a package for network-constrained biclustering of omics data

Project description
BiGAnts: network-constrained biclustering of patients and multi-omics data
ATTENTION: this package is not maintained anymore. For the same functionality please use BiCoN package
Table of contents
General info
Unsupervised learning approaches are frequently employed to identify patient subgroups and biomarkers such as disease-associated genes. Biclustering is a powerful technique often used with expression data to cluster genes along with patients. However, the genes forming biclusters are often not functionally related, complicating interpretation of the results.
To alleviate this, we developed the network-constrained biclustering approach BiGAnts which (i) restricts biclusters to functionally related genes connected in molecular interaction networks and (ii) maximizes the expression difference between two subgroups of patients.
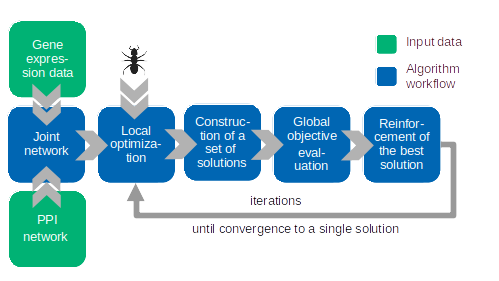
Installation
To install the package from PyPI please run:
pip install bigants
To install the package from git:
git clone https://github.com/biomedbigdata/BiGAnts-PyPI-package
python setup.py install
Data input
The algorithm needs as an input one CSV matrix with gene expression/methylation/any other numerical data and one CSV file with a network.
Please note, that even though the algorithm will accept any IDs, all visualisation tools except entrez genes IDs as an input.
Numerical data
Numerical data is accepted in the following format:
- genes as rows.
- patients as columns.
- first column - genes IDs (can be any IDs).
For instance:
| Unnamed: 0 | GSM748056 | GSM748059 | ... | GSM748278 | GSM748279 | GSM1465989 |
|---|---|---|---|---|---|---|
| 1454 | 0.053769 | 0.117412 | ... | -0.392363 | -1.870838 | -1.432554 |
| 201931 | -0.618279 | 0.278637 | ... | 0.803541 | -0.514947 | 2.361925 |
| 8761 | 0.215820 | -0.343865 | ... | 0.700430 | 0.073281 | -0.977656 |
| 2703 | -0.504701 | 1.295049 | ... | 1.861972 | 0.601808 | 0.191013 |
| 26207 | -0.626415 | -0.646977 | ... | 2.331724 | 2.339122 | -0.100924 |
There are 2 examples of gene expression datasets that can be placed in the "data" folder
- GSE30219 - a Non-Small Cell Lung Cancer dataset from GEO for patients with either adenocarcinoma or squamous cell carcinoma.
- TCGA pan-cancer dataset with patients that have luminal or basal breast cancer. Both can be found here
Network
An interaction network should be present as a CSV table with two columns that represent two interacting genes. Without a header!
For instance:
| 6416 | 2318 |
|---|---|
| 6416 | 5371 |
| 6416 | 351 |
| 6416 | 409 |
| 6416 | 5932 |
| 6416 | 1956 |
There is an example of a PPI network from BioiGRID with experimentally validated interactions here.
Main functions
Here we give a general description of the main functions provided. Please note, that all functions are annotated with dockstrings and therefore the full information can be found with help() method, e.g. help(results.save).
1.data_preprocessing(path_expr, path_net, log2 = False, size = 2000)
Parameters:
- path_to_expr: string, path to the numerical data
- path_to_net: string, path to the network file
- log2: bool, (default = False), indicates if log2 transformation should be applied to the data
- size: int, optional (default = 2000) determines the number of genes that should be pre-selected by variance for the analysis. Shouldn't be higher than 5000.
- no_zero: (default - none) indicate the fraction of allowed non-zero values for each patient. For instance no_zero = 0.8 means that all genes which have no expression for at least 80% of patients will be removed from the analysis
Returns:
- GE: pandas data frame, processed expression data
- G: networkX graph, processed network data
- labels: dict, for mapping between real genes/patients IDs and the internal ones
- rev_labels: dict, additional dictionary for mapping between real genes/patients IDs and the internal ones
- BiGAnts*(GE,G,L_g_min,L_g_max) creates a model for the given data:
Parameters:
- GE: pandas dataframe, processed expression data
- G: networkX graph, processed network data
- L_g_min: int, minimal solution subnetwork size
- L_g_max: int, maximal solution subnetwork size
Methods:
BiGAnts.run(self, n_proc = 1, K = 20, evaporation = 0.5, show_plot = False)
- K: int, default = 20, number of ants. Fewer ants - less space exploration. Usually set between 20 and 50
- n_proc: int, default = 1, number of processes that should be used(can not be more than K)
- evaporation, float, default = 0.5, the rate at which pheromone evaporates
- show_plot: bool, default = False, set true if convergence plots should be shown during the iterations
Example
Import the package:
from bigants import data_preprocessing
from bigants import BiGAnts
from bigants import results_analysis
Set the paths to the expression matrix and the PPI network:
path_expr,path_net ='/data/gse30219_lung.csv', '/data/biogrid.human.entrez.tsv'
Load and process the data:
GE,G,labels, _= data_preprocessing(path_expr, path_net)
Set the size of subnetworks:
L_g_min = 10
L_g_max = 15
Set the model and run the search:
model = BiGAnts(GE,G,L_g_min,L_g_max)
solution,scores= model.run_search()
Results analysis
BiGAnts package also allows a user to save the results and perform an initial analysis.
The examples below show the basic usage, for more details please use python help() method, e.g. help(results.save).
- First, the object for results analysis must be created:
results = results_analysis(solution, labels)
This will allow to easily access the resulting biclusters and their initial IDs as well as perform a more complicated analysis.
To access IDs of patients in the first bicluster:
results.patients1
To access IDs of genes IDs in the first bicluster:
results.genes1
Same logic applies to the second bicluster.
If in the further analysis you would like to use gene names, please set 'convert' to True and specify the original gene IDs, i.e.:
results = results_analysis(solution, labels, convert = True, origID = 'entrezgene')
Some other options for the original gene ID: ensembl.gene', 'symbol', 'refseq', 'unigene', etc For all possibe option please check the reference for MyGene.info gene query web service
- To save the solution:
results.save(output = "results/results.csv")
- Visualise the resulting networks colored with respect to their difference in expression patterns in patients clusters:
results.show_networks(GE, G, output = "results/network.png")
- Visualise a clustermap of the achieved solution alone or also along with the known patients' groups. Just with the BiGAnts results:
results.show_clustermap(GE, G, solution, labels, output = "results/clustermap.png")
If you have a patient's phenotype you would like to use for comparison, please make sure that patients IDs are exactly (!) matching the IDs that were used as an input. The IDs should be represented as a list of two lists, e.g.:
true_classes = ['GSM748056', 'GSM748059',..], ['GSM748278', 'GSM748279', 'GSM1465989']
results.show_clustermap(GE, G, solution, labels, output = "results/clustermap.png", true_labels = true_classes)
- Given a known phenotype in a format described above, BiGAnts can also return Jaccard index of the achieved patients clustering with a given phenotype:
results.jaccard_index(true_labels = true_classes)
results.enrichment_analysis(solution, labels, library = 'GO_Biological_Process_2018', "results")
After the execution of the given above code, in the /results directory a user can find a table with enriched pathways as well as enrichment plots. Other available libraries can be used as well, e.g. 'GO_Molecular_Function_2018' and 'GO_Cellular_Component_2018'. In total there are 159 libraries available at the moment and the full list can be found by typing:
import gseapy
gseapy.get_library_name()
Quality control
Algorithm convergence
The best way to check if the algorithm produced high-quality results and there are no issues with the parameters is to analyse the convergence plot:
results.convergence_plot(scores)
- The algorithm converged:
- If the maximum score has stabilised for several iterations in a row (default is 6).
OR
- If the average score became equal (or nearly equal) to the maximal score:
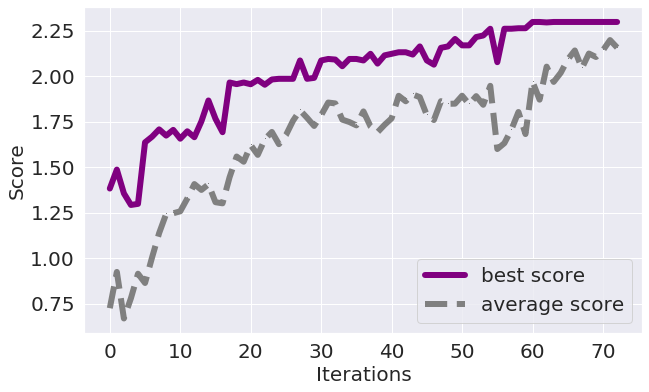
- The algorithm did not converge:
If the average and the maximal score improve over the iterations but do not stabilize then just increase the number of maximally allowed iterations:
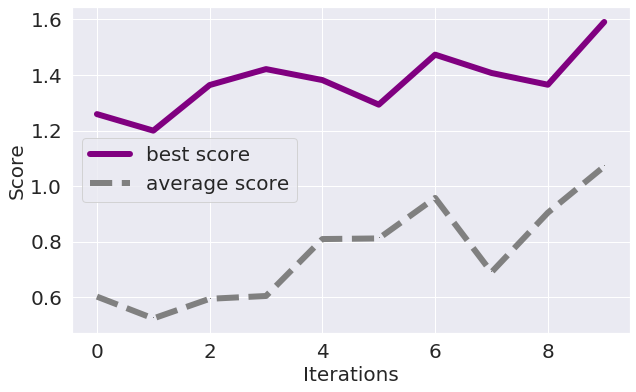
If the scores do not stabilize even after 60-100 iterations, please contact us.
Bad probability update
If you got the following error message:
AssertionError: bad probability update
It can mean one of the following issues:
- The setting of the algorithm is way to restrictive for your problem. You can try to fix it by repeating the analysis with th = 0, or even th = -1 e.g.:
model = BiGAnts(GE,G,L_g_min,L_g_max)
solution,scores= model.run_search(th = 0)
- Otherwise, the problem might be related to the way you have processed your data. Please make sure that you do not have not expressed genes for the magority of the patients, that you log2 or even log10 scaled your values.
Cite
BiGants was developed by the Big Data in BioMedicine group and Computational Systems Medicine group at Chair of Experimental Bioinformatics.
If you use BiGAnts in your research, we kindly ask you to cite the following manuscript:
Lazareva, O., Van Do, H., Canzar, S., Yuan, K., Baumbach, J., Kacprowski, T., List, M.: BiGAnts: Network-constrained biclustering of patients and omics data. [Submitted]
Contact
If you have difficulties using BiGAnts, please open an issue at out GitHub repository. Alternatevely, you can write an email to:
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file bigants-2.2.2.tar.gz.
File metadata
- Download URL: bigants-2.2.2.tar.gz
- Upload date:
- Size: 22.6 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/1.13.0 pkginfo/1.5.0.1 requests/2.22.0 setuptools/41.2.0 requests-toolbelt/0.9.1 tqdm/4.32.1 CPython/3.7.3
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
00fed958d3e0fca6422daeb0d475370ba461f2ae1713201fdef50cc7ddfddead
|
|
| MD5 |
0b881abab48edf9c1d789051536224e5
|
|
| BLAKE2b-256 |
13c1219851740075599f15c233351753c4c20c0c89156311d5975036f003651c
|







