Generate macrocomplexes superimposing paired interacting elements.

Project description

Complex Constructor
Generate macrocomplexes superimposing paired interacting elements
protein - protein or protein - DNA
the result is stored in a PDB file
Table of Content
Why building complexes?
The total number of proteins in humans is around 20K. From this quantity we already know the structure of 4K proteins. However, for 6K we have good templates and for other 6K of them, we have reasonably good templates; that means we have more or less 50-70% of the structure of human proteins covered.
On the other hand, the number of interactions that may occur between all the proteins is still unknown. There are some studies estimating 130-650K interactions, while others estimate more than millions. In this context, 120K interactions are confirmed from experiments and only 7K structures of complexes are known, which is a very small proportion of the total number of possible interactions anyway.
So, the complex-structure coverage, including homologs, is around the 30%, much less than the mentioned coverage of monomers. That means there is still a long way to go into the complex-structure study field.
The 3D structure of interacting proteins is necessary for them to develop their function. It is also fundamental for molecular recognition and contributes to the complexity of protein interaction networks. However, identifying the structure of interacting proteins forming complexes, is not an easy task. In monomers, homology modelling can be used to nearly cover all folds to determine a protein structure but, for interactions that is not enough.
Complex Constructor tries to generate macrocomplex structures. To do so, the superimposing technique is used: it receives a list of pdb files, each of these files contains the structure of an interacting pair and, by superimposing the common elements of different pairs, it builds the final structure. Although there are other methods to generate macrocomplexes, the superimposition is fast and effective. Furthermore, not only protein-protein interacting pairs can be analyzed, but also proteins with DNA, to end up generating a macrocomplex structure of proteins and DNA chains.
The core of Complex Constructor is the constructor function, which is the responsible of the building process. The pdbs of the interacting pairs are analyzed and classified using the information of the FASTA file. The common chains in different pdb files are identified: they will have more than 99% of identity in their sequences. After this classification, constructor begins to append elements to the macrocomplex: it starts with a pair, then it looks for another pair that has a common chain with the starting one and, if after superimposing the common chain, the structure has not clashed, it appends the new chain. Like this, the macrocomplex has now three chains. In the next step, the process will be repeated by looking for another pair, superimposing the common chain and checking the possible clashes. The program is described here.
In addition, as a lot of complexes follow a determined stoichiometry, Complex Constructor can also make the construction following it. If a stoichiometry is defined, the final structure will contain the exact number of elements indicated by it. In other case, the structure will contain each different chain only once.
Finally, a new pdb file with the resulting structure of the macrocomplex is stored in the output folder called outputFolder_model.pdb.
Installation
Prerequisites:
- Python 3.0
https://www.python.org/download/releases/3.0/
There are two different ways to download the package: using pip3 or cloning the repo.
Option 1. Install from pip
- Install Complex Constructor with the Python package installer
pip3
$ pip3 install complexconstructor
Option 2. Clone
- Otherwise, you can clone the repo to your local machine
$ git clone https://github.com/Argonvi/SBI-PYT_Project.git
- Install the package
$ cd SBI-PYT_Project
$ sudo python3 setup.py install
Setup
Complex Constructor requires the package BioPython https://biopython.org/wiki/Download
- The installation is easy using Python package management tool
pip3
$ pip3 install biopython
$ pip3 install biopython --upgrade
Download the data to run the examples
To run the different examples of this tutorial, download the folder examples. Inside it you can find all the required inputs to execute the examples.
You can download the compressed folder with the following command:
$ wget https://github.com/Argonvi/SBI-PYT_Project/raw/master/examples.tar.gz
Then uncompress with:
$ tar xvfz examples.tar.gz
Options
Complex Constructor can be run using command-line arguments or using the graphical interface.
Command-line
You can introduce the different arguments via command-line:
Mandatory arguments
To execute Complex Constructor three arguments are required:
-
-fa--fasta: FASTA file with the sequences of the proteins or DNA that will conform the complex. -
-pdb--pdbDir: directory containing the pdb files with the structure of the pairs that will conform the complex. -
-o--output: name of the directory where the complex results will be stored.
Note that, if the output directory already exists, the results will be overwritten.
Optional arguments
-
-v--verbose: show the detailed progression of the building process in a file called 'ComplexConstructor.log'. It will be stored in the output directory. -
-st--stoichiometry: name of the file in .txt format containing the stoichiometry of the complex. The information of the stoichiometry must be in the following order: the ID of the sequence chain (concordant with the FASTA file ID) followed by ':' and the number of times it has to be present in the complex.ID_FASTA_file:stoichiometryOne per line.In case this option is not used, the program will build the complex using each chain only once.
Take a look at some examples here.
Graphical interface
Otherwise, the macrocomplex can also be built using the graphical interface:
$ cconstruct -gui
In this case just the
-guitag is needed!
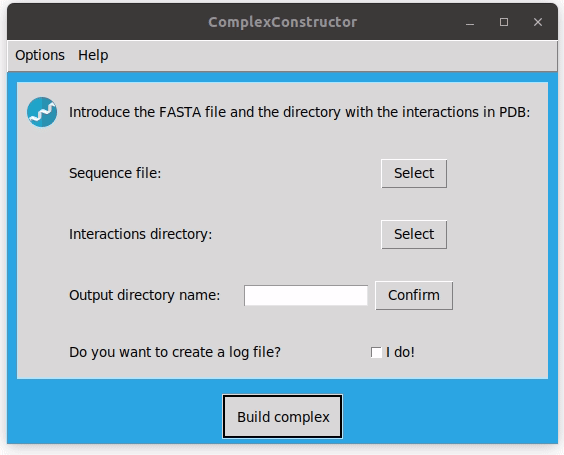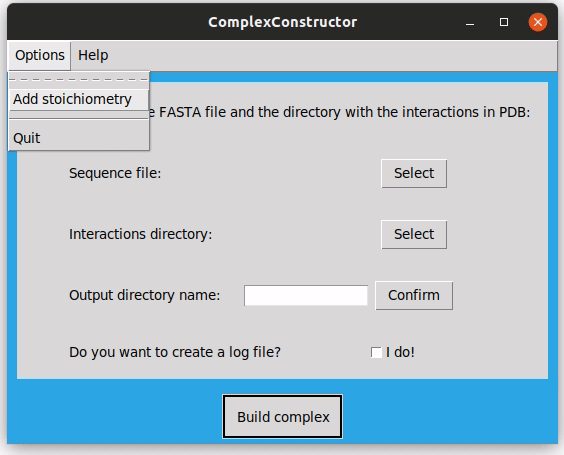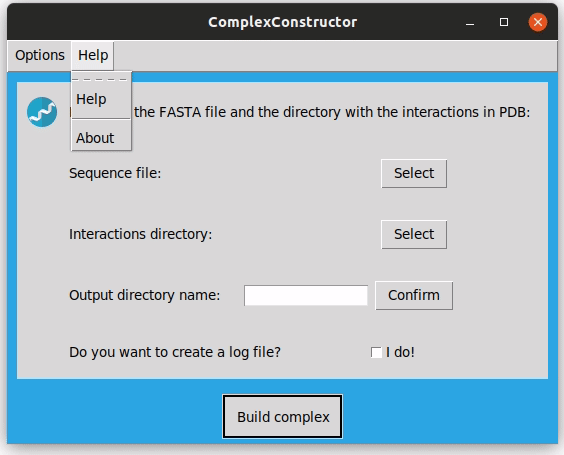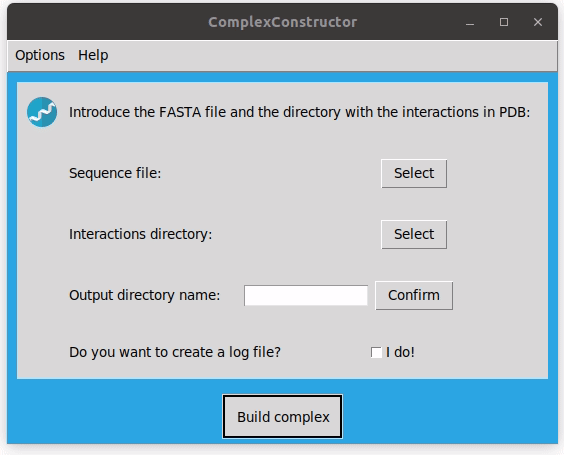
- To build the macrocomplex fill in the main window requirements. As for running the program via command-line, a FASTA file with the sequences and a directory with the pdb files of the interacting pairs are required. In addition, a name for the folder where the results will be stored is needed, after typing it you should confirm it.
Note that, to select the PDB directory you have to enter in the desired directory and then select it. You can see how it works in the examples.
Furthermore, additional options can be set:
-
In the main window you can specify if you want to create a log file where the process of the execution will be displayed. It will be stored in the output directory.
-
In the top menu, in the 'Options' dropdown, there is the 'Add Stoichiometry' option. You can upload a file with a determined stoichiometry to be applied to the macrocomplex. As said before, the format of this file has to be the ID of the sequence, concordant with the one in the FASTA file, followed by ':' and a number. This number will be the number of times the corresponding sequence will be in the final complex.
Finally, in the top menu you can consult the Complex Constructor 'Help' as well.
Examples
As said before, to generate any macrocomplex structure it is required the FASTA file and a direcory with paired structures in pdb format. All the data needed to execute the following examples is in the folder examples, remember that you have to download it as described in the installation section.
- The output folder will be created in the 'current' directory, the one from which you execute the Complex Constructor. Our recommendation to follow the following examples is to create a working directory, for example
ComplexConstructorModels, and place theexamplesdirectory inside it,ComplexConstructorModels/examples. Now navigate using thecdcommand to reachComplexConstructorModelsdirectory.
$ cd ComplexConstructorModels
Like this, the output folders will be stored in this directory.
Inside the folder
examplesthere are all the examples we will describe in this tutorial in the corresponding directories:1gzx,5fj8, etc. Each of them contains the required files to run Complex Constructor and also a pdb file of the reference structure,exampleRef.pdb, in order to check the superimposition between the constructed model and the real structure.
1GZX
Let's begin with the first example, the protein 1GZX. It is a small complex composed by two different amino acid chains and each one of them appears two times (stoichiometry 2A2B), so the final structure has four chains. In order to perform the construction of T state haemoglobin (1GZX), we need a .txt file where the stoichiometry is specified. This file can be found in the folder examples/1gzx and it is called 1gzx_st.txt:
1GZXa:2
1GZXb:2
- Command line execution:
$ cconstruct -fa examples/1gzx/1gzx.fa -pdb examples/1gzx/1gzxDir -o 1GZX -st examples/1gzx/1gzx_st.txt -v
-fa, mandatory: followed by the location of the FASTA file1gzx.fa.
This file contains two IDs followed by the corresponding sequence, e.g.
1GZXa,1GZXb. Note that, the IDs in1gzx_st.txthave to be concordant with them.
-pdb, mandatory: followed by the location of the directory with paired structures in pdb, that is1gzxDir.
In this case inside this folder we should have at least three pdb files, e.g.
1gzx_AB.pdb,1gzx_AC.pdb,1gzx_AD.pdb. If there are redundant pairs they won't be considered.
-o, mandatory: followed by the name of the output directory where the results will be stored,1GZX.
If a directory with the same name already exists in the working folder it will be overwritten.
-
-st: followed by the location of the file that contains the stoichiometry information, that is, the file1gzx_st.txt. -
-v: when it is used it turns ON the verbose option. It is always recommended to create a logfile where the process information will be displayed. In order to deactivate the creation of the logfile, do not add the-vflag.
- Graphical interface execution:
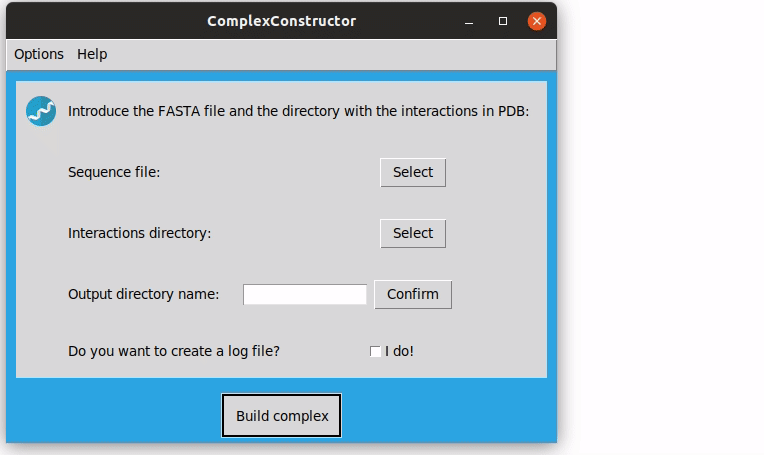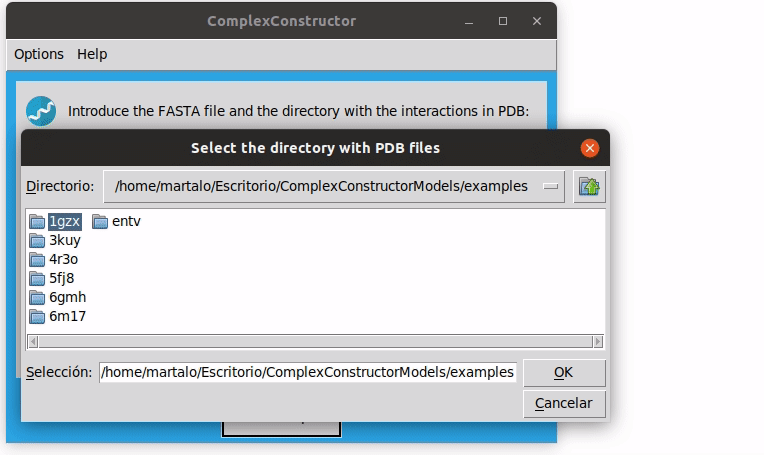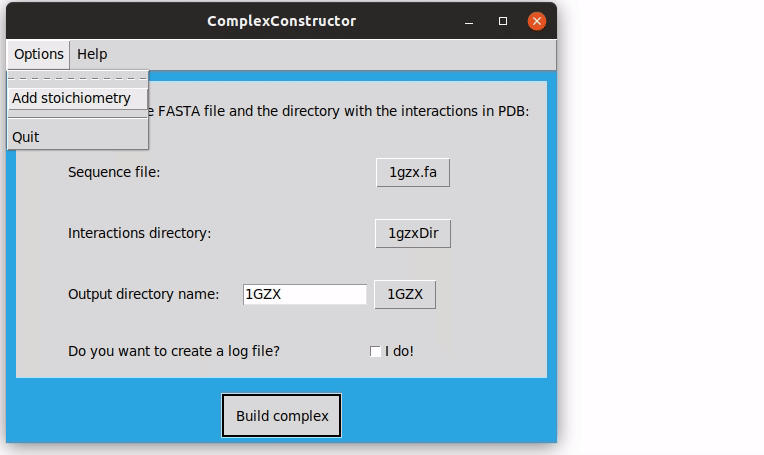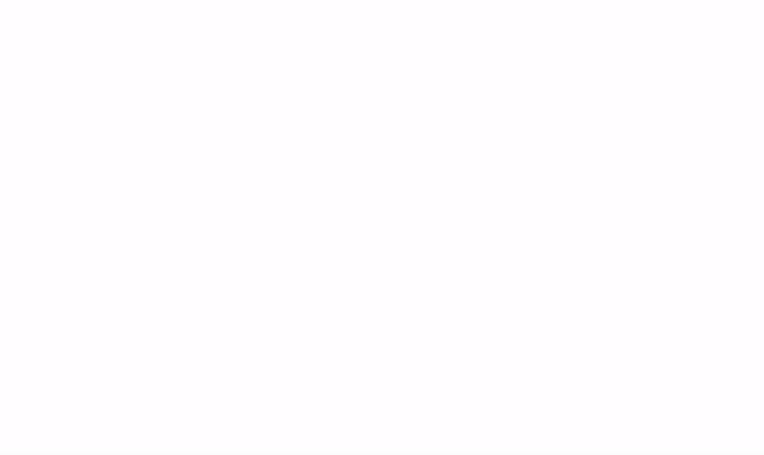
The resulting structure is stored in the current directory, ComplexConstructorModels, in the folder 1GZX, file 1GZX_model.pdb.
| Complex Constructor | Reference structure | Superimposition |
|---|---|---|
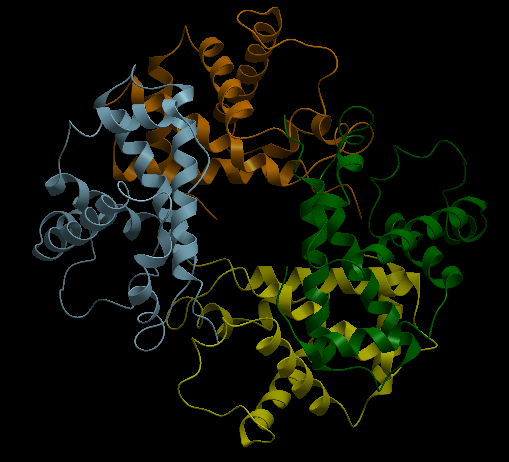 |
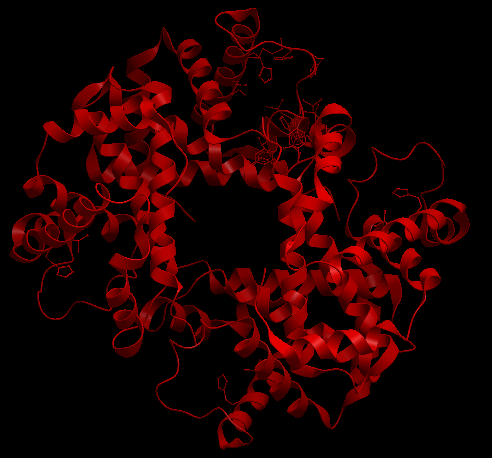 |
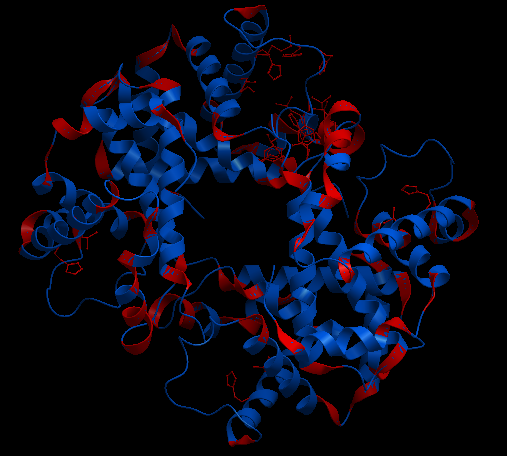 |
In the superimposition image we can observe the reference structure in red and the structure obtained with Complex Constructor in blue. We observe that the colours are mixed in most of the chains as our model fits the reference downloaded from PDB quite well.
Using Chimera we computed the RMSD between 146 pruned atom pairs and obtained a result of 0.000 angstroms.
3KUY
3KUY is a complex composed by a DNA coil and a core made of protein chains. There are four different amino acid chains and one nucleotide chain, all of them have stoichiometry two, making a total of 10 chains. The procedure to run this example is the same as the explained before. The data to construct the complex is inside the folder examples/3kuy. Execution with command-line arguments:
$ cconstruct -fa examples/3kuy/3kuy.fa -pdb examples/3kuy/3kuyDir -o 3KUY -st examples/3kuy/3kuy_st.txt -v
The resulting structure is stored in the file 3KUY_model.pdb, which is in the directory 3KUY.
In order to construct this complex (and all the following examples) using the graphical interface, we should repeat the same process as the one explained in the previous example, but in this case, we will take the inputs from the folder
examples/3kuy.
| Complex Constructor | Reference structure | Superimposition |
|---|---|---|
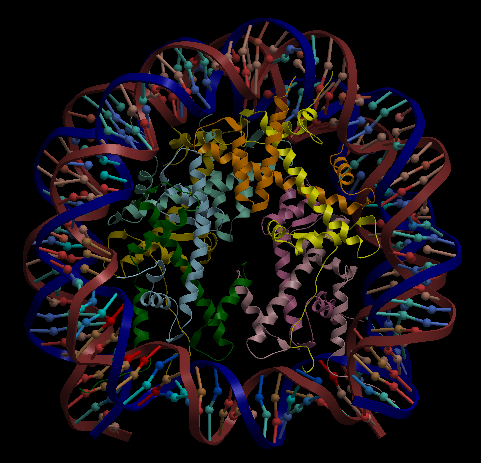 |
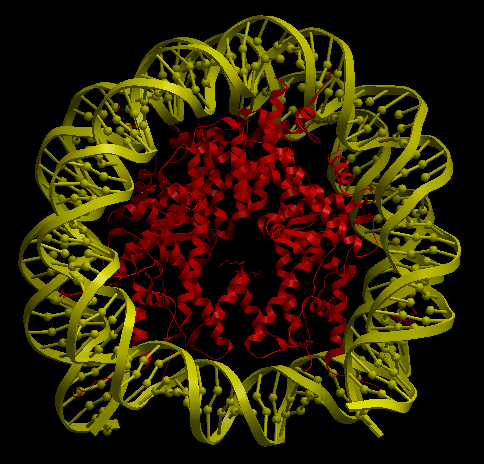 |
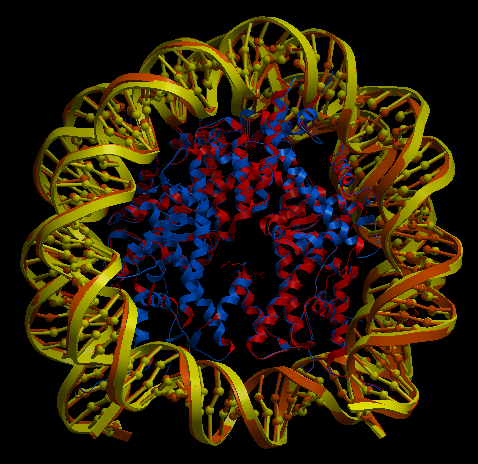 |
In the superimposition image we can observe the amino acid chains of the reference structure in red and the DNA chains in yellow. The colours of the structure obtained with Complex Constructor are blue and orange respectively.
We observe that the whole complex is correctly constructed and after superimposing the obtained structure with the structure downloaded from PDB, we can see that both protein chains and DNA chains fit quite well with the reference structure.
The RMSD computed with Chimera between 106 pruned atom pairs is 0.000 angstroms.
4R3O
The Human 20S Proteasome (4R3O), is a bigger complex but just made of amino acid chains. It is symmetric and it is composed by 14 different chains, all of them with stoichiometry two, making a total of 28 chains in the complex. Its input data can be found in examples/4r3o. Execution with command-line arguments:
$ cconstruc -fa examples/4r3o/4r3o.fa -pdb examples/4r3o/4r3oDir -o 4R3O -st examples/4r3o/4r3o_st.txt -v
The resulting structure is stored in the file 4R3O_model.pdb, which is in the directory 4R3O.
| Complex Constructor | Reference structure | Superimposition |
|---|---|---|
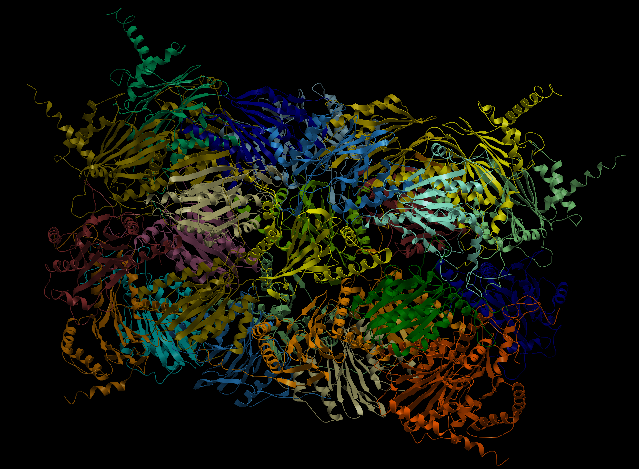 |
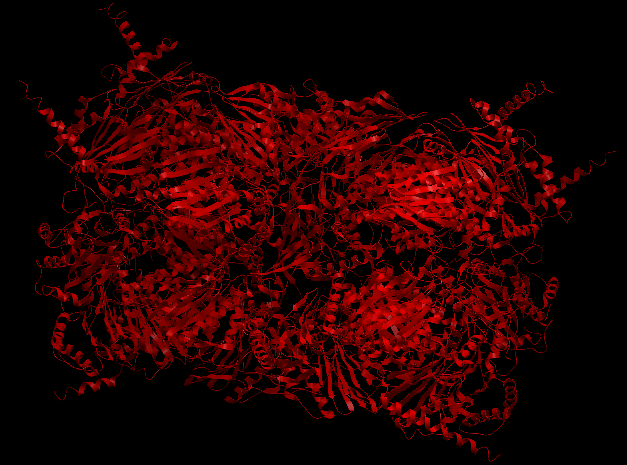 |
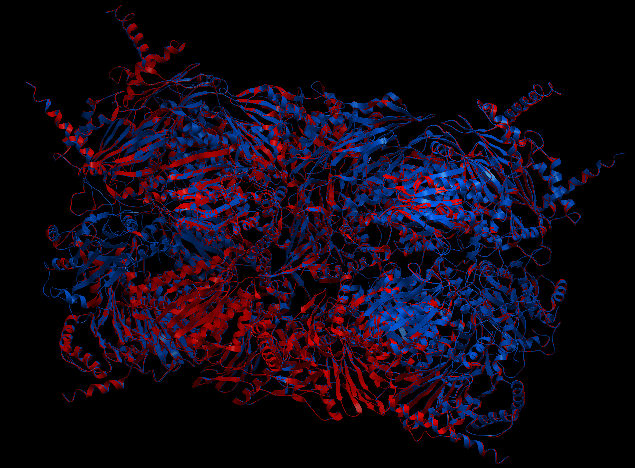 |
In this case, as well, the constructed model fits the reference structure.
The RMSD between 250 pruned atom pairs is 0.000 angstroms.
5FJ8
The next complex is composed by amino acid and nucleotide sequences but, in this case, it is not symmetric. It is composed by 20 different chains and all of them are present just once in the structure. The required inputs for the construction are in examples/5fj8.
$ ccontruct -fa examples/5fj8/5fj8.fa -pdb examples/5fj8/5fj8Dir -o 5FJ8 -st examples/5fj8/5fj8_st.txt -v
The resulting structure is stored in the file 5FJ8_model.pdb, which is in the directory 5FJ8.
| Complex Constructor | Reference structure | Superimposition |
|---|---|---|
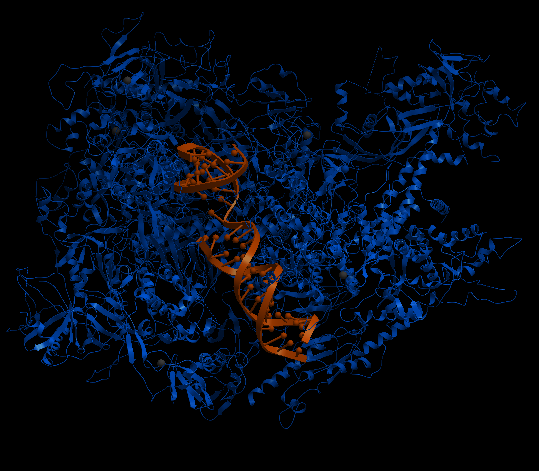 |
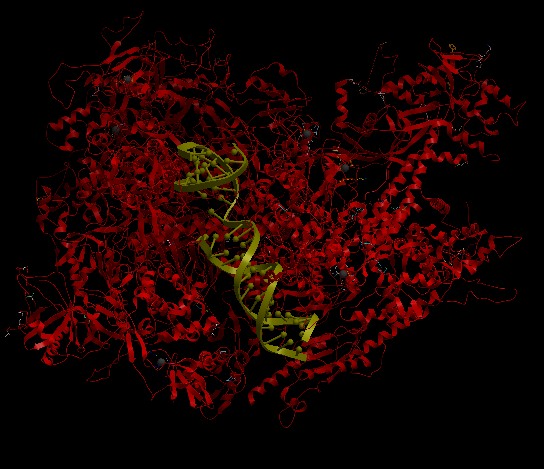 |
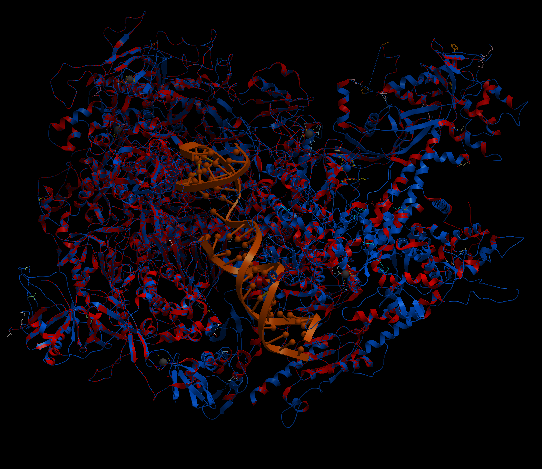 |
The created model and the reference structure are superimposed quite well as in previous cases. In this particular case, the amino acid chain Q, had several amino acids labelled as 'unknown' in the pdb files and as 'X' in the FASTA sequence. To deal with this, we had to take out these 35 amino acids from input files (FASTA file and pdb files), and so, the Q chain is partly constructed in the model. Nevertheless, the rest of the structure is correctly reproduced.
The RMSD between 1422 pruned atom pairs is 0.000 angstroms.
6GMH
Another non-symmetric example, but also with DNA sequences, is 6GMH. It has 17 amino acid chains and 3 DNA chains and as in the previous case, all of them appear only once in the structure. The required inputs for the construction are in examples/6gmh.
$ cconstruct -fa examples/6gmh/6gmh.fa -pdb examples/6gmh/6gmhDir -o 6GMH -st examples/6gmh/6gmh_st.txt -v
The resulting structure is stored in the file 6GMH_model.pdb, which is in the directory 6GMH.
| Complex Constructor | Reference structure | Superimposition |
|---|---|---|
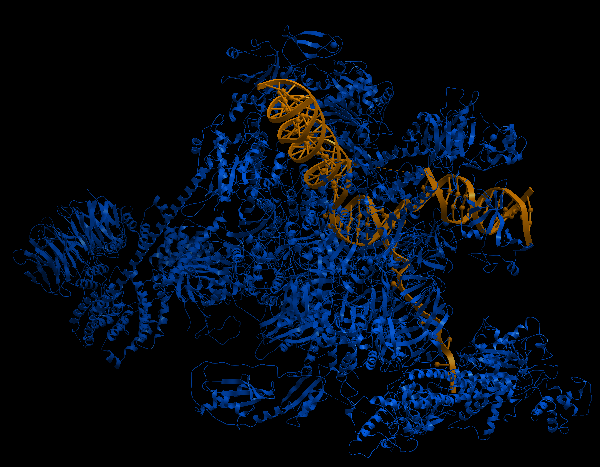 |
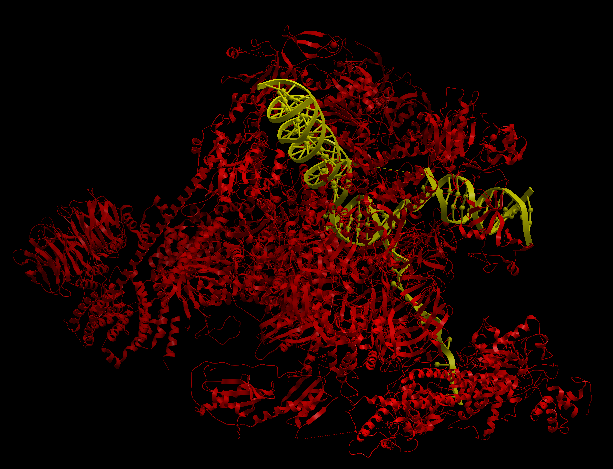 |
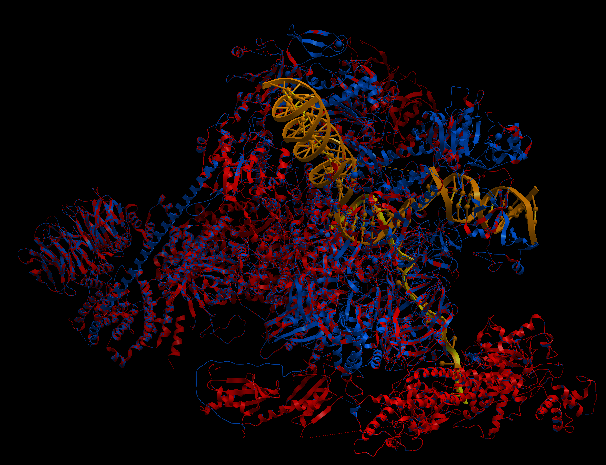 |
In this case, the pdb structure had bigger regions labelled as 'unknown'. As those regions are not recognized by Complex Constructor, we removed them from the input pdb pairs and from the FASTA sequences, as in the previous example. In the Q chain, from a total of 884 amino acids, 300 of them were labelled as 'unknown', so only 584 remained in order to construct the model, (that is why this chain is not completely constructed in our model). It happened to be the same in chain M, but in this case only 8 amino acids were missing. On the other hand, the chains U, V and X had to be completely deleted in our model, since all their amino acid were labelled as 'unknown' in the pdb file.
Even though we had to remove several amino acids and some chains, the model is well constructed and it fits the reference structure as in previous examples, (as we can see in the image of the superimposition).
The RMSD between 1441 pruned atom pairs is 0.000 angstroms.
6M17
We have tried to build a newer complex such as 6m17, which is a membrane protein of the SARS coronavirus. It has been released at PDB on march 2020. Knowing the structure of this membrane protein and how it interacts with other proteins can be very useful in order to find a vaccine or a drug that prevents us from this virus. It has three different amino acid chains and all of them have stoichiometry two, making a total of 6 chains. The data to construct the complex is inside the folder examples/6m17. Execution with command-line arguments:
$ cconstruct -fa examples/6m17/6m17.fa -pdb examples/6m17/6m17Dir -o 6M17 -st examples/6m17/6m17_st.txt -v
The resulting structure is stored in the file 6M17_model.pdb, which is in the directory 6M17.
| Complex Constructor | Reference structure | Superimposition |
|---|---|---|
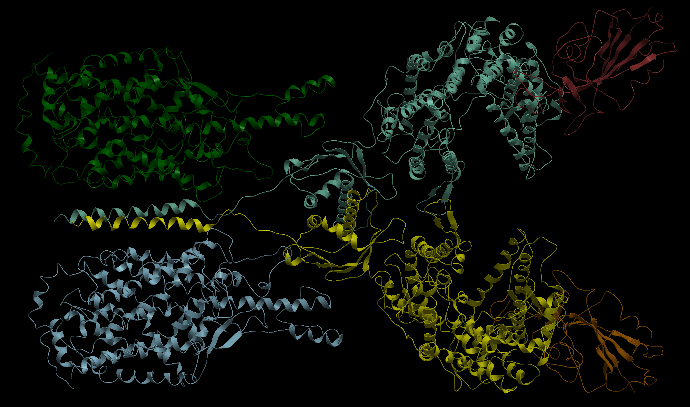 |
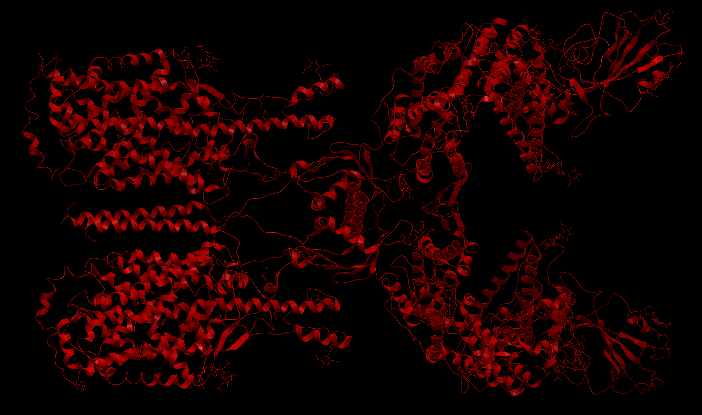 |
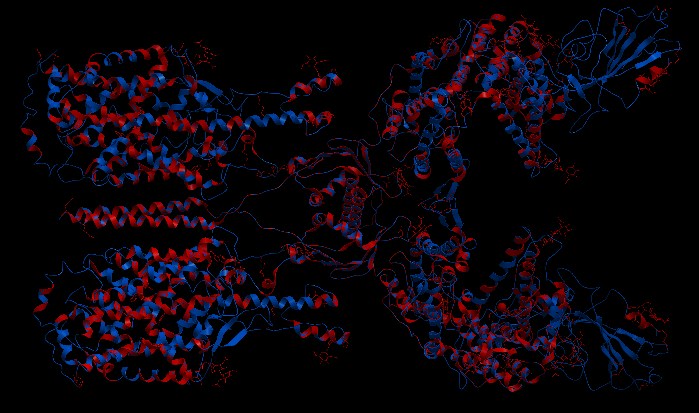 |
We observe that the whole complex is correctly constructed and after superimposing the obtained structure with the structure from PDB, we can see that all the chains fit quite well with the reference structure.
The RMSD computed with Chimera between 748 pruned atom pairs is 0.000 angstroms.
Enterovirus capsid
In order to see how the Complex Constructor works with a complex whose structure is unknown, we have run Complex Constructor with an enterovirus capsid, composed by three different amino acid chains. To execute this example, we provide a stoichiometry file with stoichiometry equal to 18 for all three chains. However, with a total of 54 chains the complex is not totally constructed.
The following image shows the construction of the complex with stoichiometry 32, a total of 96 chains. To perform the construction with so many chains, we had to modify the script: the program was able to generate a total of 54 chains, with letters A-Z and a-z, so the maximum stoichiometry for three chains was 18. In addition to this limitation, ICM and Chimera, allow a maximum of 99999 atoms to be represented, and the complex exceeded this number.
To solve both issues, we made modifications for this particular case so that we could create two different pdb files, like this, we divided the pdb structure in two files ENTV_model_part1.pdb and ENTV_model_part2.pdb, to be able to repeat the chain letters in the second file and open both files with ICM or Chimera. These two pdb files are included in the directory examples/entv.
Nevertheless, as the modifications were just performed to build this particular case, the stoichiometry file that we have included to run this example, is the one that will work with the 'default' script, this is, stoichiometry equal to 18 for all the three chains. Like this, the resulting pdb file will be partly constructed. To run the enterovirus capside 'reduced' example use the following commands:
$ python3 complexconstructor -fa examples/entv/entv.fa -pdb examples/entv/entvDir -o ENTV -st examples/entv/entv_st.txt -v
The resulting structure is in directory ENTV, in the file ENTV_model.pdb, with the 'reduced' structure.
The 'extended' structure, contained in ENTV_model_part1.pdb and ENTV_model_part2.pdb, is as follows:

We can see that, the structure is not completed yet, but using a third pbd would allow adding more chains and complete the complex. The structure of the capside is correctly visualized.
Performance
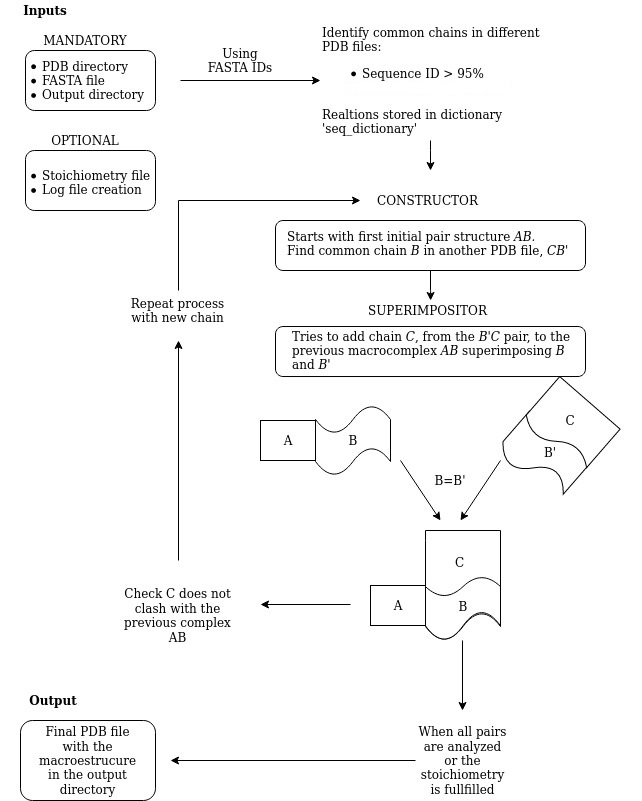
- Before adding a new chain to the macrocomplex, the number of clashes between the new chain and the previous structure is checked. The function
sequence_clashingcalculates how many CA, P or N1 atoms from the new chain are closer than 2 angstroms to any other CA, P or N1 atom of the previous macrocomplex, this is, the number of clashes. If the number of clashes is above 35, the new chain won't be added to the macrocomplex.
Structure of the package
ComplexConstructor
-
main.py: main module of Complex Constructor. It connects with all the rest of modules. -
utilities.py: module with all the functions needed to run the construction of macrocomplexes, as theconstructorfunction orsuperimpositor. -
argparser.py: reads and organizes the command-line arguments. -
interface.py: contains the configuration of the graphical interface and passes the inputs to the main module. -
logProgress.py: this module creates the log file and its contents when the verbose option is selected. -
helpText.py: it is a module with the help text to be added to the graphical interface 'Help' option.
Limitations
-
The main limitation of the program is the preparation of the input files. The FASTA file must have each ID just once and the sequences must be unique, that means, if a structure has a stoichiometry 2A2B, the FASTA file should contain just once the sequence of the chain A and once the sequence of the chain B.
-
Furthermore, as we have set that the sequence identity should be over 99%, the sequences in the pdb files and those in the FASTA file should be almost identical in order to be identified as the same sequence. Nevertheless, it is not a big limitation as it is a parameter that could be changed in the code if needed.
-
The amino acids labelled as 'unknown' in the pdb files and as 'X' in the FASTA sequences, cannot be correctly identified so they should not be present in the input files. As a consequence, those chains containing unknown amino acids are just partly constructed in the final model.
-
We would have also liked to check the Z-score for the macrocomplexes with unknown structure through the energy analysis. We thought about creating a .cmd file to be executed with ProSa 2003 in order to create a file with the z-score table. Like this, we would have been able to check if the structure was energetically valid. However, as the Prosa software is temporarily inaccessible we have left this part of the analysis for the moment.
Team
| Paula Gomis Rosa | Arturo González Vilanova | Marta López Balastegui |
|---|---|---|
 |
 |
 |


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file complexconstructor-0.1.6.tar.gz.
File metadata
- Download URL: complexconstructor-0.1.6.tar.gz
- Upload date:
- Size: 92.1 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.1.1 pkginfo/1.5.0.1 requests/2.23.0 setuptools/46.1.1 requests-toolbelt/0.9.1 tqdm/4.43.0 CPython/3.6.9
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
f6db57fa9b29a8921d28cf4bc5d0f1bbf202bbb458a2ddeef71758a6e48695c4
|
|
| MD5 |
6018444101745ecc4ba9ae8088ce76a1
|
|
| BLAKE2b-256 |
1d6a3d98c45cb65c4faca8cfb4791cfa42f05d387ca256bd6ecf537698cd0010
|
File details
Details for the file complexconstructor-0.1.6-py3.6.egg.
File metadata
- Download URL: complexconstructor-0.1.6-py3.6.egg
- Upload date:
- Size: 97.8 kB
- Tags: Egg
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.1.1 pkginfo/1.5.0.1 requests/2.23.0 setuptools/46.1.1 requests-toolbelt/0.9.1 tqdm/4.43.0 CPython/3.6.9
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
3e7244717ea4037383354ac33ae7acbed6fb80b97be92300869503e82e61cd33
|
|
| MD5 |
821a052cd2e00d6b23826e5cb7cf3787
|
|
| BLAKE2b-256 |
022c91456bf8f2f32ee73d6ccf5d7363f8a7c8669c23006930bd7088fad6bbef
|







