Scalable and reliable demulitplexing for single-cell RNA sequencing.

Project description



Demuxalot
Reliable and efficient identification of genotypes for individual cells in RNA sequencing. Demuxalot refines its knowledge about genotypes directly from the data.
Demuxalot is fast and optimized to work with lots of genotypes, enabling efficient reutilization of inferred information from the data.
Preprint is available at biorxiv.
Background
During single-cell RNA-sequencing (scRnaSeq) we pool cells from different donors and process them together.
- Pro: all cells come through the same pipeline, so preparation/biological variation effects are cancelled out from analysis automatically. Also experiments are much cheaper!
- Con: we don't know cell origin, everything is mixed!
Demuxalot solves the con: it guesses genotype of each cell by matching reads coming from cell against genotypes. This is called demultiplexing.
Comparisons
Demuxalot shows high reliability, data efficiency and speed. Below is a benchmark on PMBC data with 32 donors from preprint
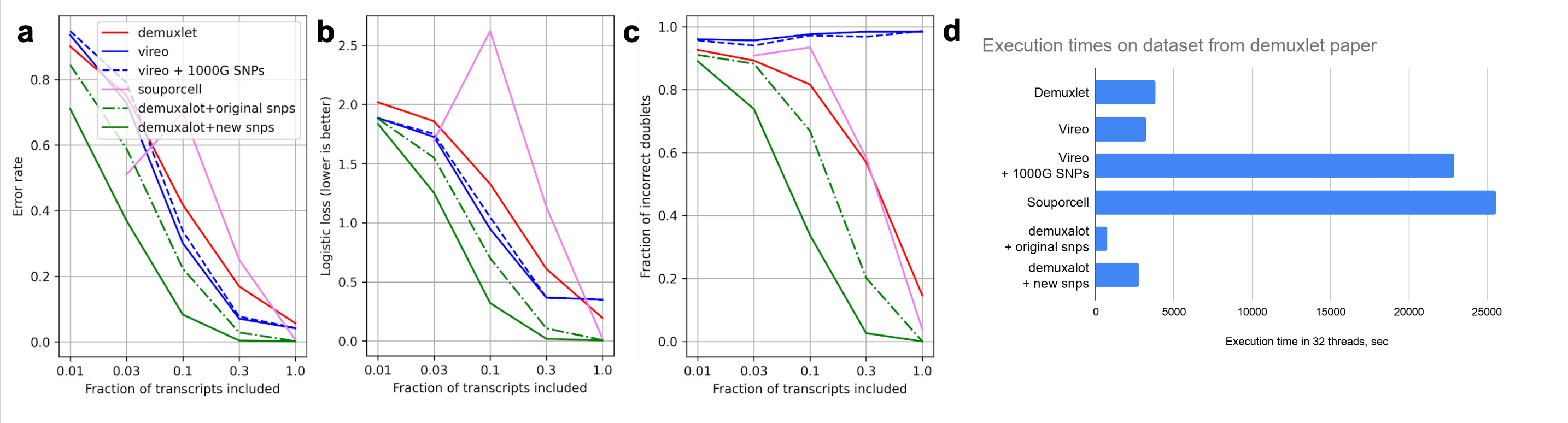
[!NOTE] we used
demuxalotinternally for a number of challenging scenarios with a large biobank and low-depth sequencing, and it shines in these scenarios too. Actually, that's why algorithm was created in the first place.
Known genotypes and refined genotypes: the tale of two scenarios
Typical approach to get genotype-specific mutations are
- whole-genome sequencing (expensive, very good)
- you have information about all (ok, >90%) the genotype, and it is unlikely that you need to refine it
- so you just go straight to demultiplexing
- demuxlet solves this case
- Bead arrays (aka SNP arrays aka DNA microarrays) are super cheap and practically more relevant
- you get information about ~650k most common SNPs, and that's only a small fraction, but you also pay very little
- this case is covered by
demuxalot(this package) - Illumina's video about this technology
Why is it worth refining genotypes?
SNP array provides up to ~650k positions in the genome. Around 20-30% of them would be specific for a genotype (i.e. deviate from majority).
Each genotype has around 10 times more SNV (single nucleotide variations) that are not captured by array. Some of these missing SNPs are very valuable for demultiplexing.
What's special power of demuxalot?
- much better handling of multiple reads coming from the same UMI (i.e. same transcript)
demuxalotefficiently combines information from multiple reads with same UMI and cross-checks it
- default settings are CellRanger-specific (that is - optimized for 10X pipeline). Cellranger's and STAR's flags in BAM break some common conventions, but we can still efficiently use them (by using filtering callbacks)
- ability to refine genotypes. without failing and diverging
- Vireo is a tool that was created with similar purposes. But it either diverges or does not learn better genotypes
- optimized variant calling. It's also faster than
demuxletdue to multiprocessing - this is not a command-line tool, and not meant to be
- write python code, this gives full control and flexibility of demultiplexing
Installation
Plain and simple:
pip install demuxalot # Requires python >= 3.8
Here are some common scenarios and how they are implemented in demuxalot.
Also visit examples/ folder
Running (simple scenario)
Only using provided genotypes
from demuxalot import Demultiplexer, BarcodeHandler, ProbabilisticGenotypes, count_snps
# Loading genotypes
genotypes = ProbabilisticGenotypes(genotype_names=['Donor1', 'Donor2', 'Donor3'])
genotypes.add_vcf('path/to/genotypes.vcf')
# Loading barcodes
barcode_handler = BarcodeHandler.from_file('path/to/barcodes.csv')
snps = count_snps(
bamfile_location='path/to/sorted_alignments.bam',
chromosome2positions=genotypes.get_chromosome2positions(),
barcode_handler=barcode_handler,
)
# returns two dataframes with likelihoods and posterior probabilities
likelihoods, posterior_probabilities = Demultiplexer.predict_posteriors(
snps,
genotypes=genotypes,
barcode_handler=barcode_handler,
)
Running (complex scenario)
Refinement of known genotypes is shown in a notebook, see examples/
Saving/loading genotypes
# You can always export learnt genotypes to be used later
refined_genotypes.save_betas('learnt_genotypes.parquet')
refined_genotypes = ProbabilisticGenotypes(genotype_names= <list which genotypes to load here>)
refined_genotypes.add_prior_betas('learnt_genotypes.parquet')
Re-saving VCF genotypes with betas (recommended)
Loading of internal parquet-based format is much faster than parsing/validating VCF. Makes sense to export VCF to internal format in two cases:
- when you plan to load it many times.
- when you want to 'accumulate' inferred information about genotypes from multiple scnraseq runs
genotypes = ProbabilisticGenotypes(genotype_names=['Donor1', 'Donor2', 'Donor3'])
genotypes.add_vcf('path/to/genotypes.vcf')
genotypes.save_betas('learnt_genotypes.parquet')
# later you can use it.
genotypes = ProbabilisticGenotypes(genotype_names=['Donor1', 'Donor2', 'Donor3'])
genotypes.add_prior_betas('learnt_genotypes.parquet')
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file demuxalot-0.4.3.tar.gz.
File metadata
- Download URL: demuxalot-0.4.3.tar.gz
- Upload date:
- Size: 23.9 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: python-httpx/0.28.1
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d42f507d53dddc04c7b2cc5bfd12c4947f3204d56c89c214da289d775b3ef216
|
|
| MD5 |
c5b74ee06a01ce04075eccc988869026
|
|
| BLAKE2b-256 |
c42dfccd9a1782f39757e4fe630d7eaefebb8993c9409ae0e64f7d1dc94ee71b
|
File details
Details for the file demuxalot-0.4.3-py3-none-any.whl.
File metadata
- Download URL: demuxalot-0.4.3-py3-none-any.whl
- Upload date:
- Size: 28.5 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: python-httpx/0.28.1
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
1a1765c0f39fc4b90efe0c65f2d11253937eacdbc469e8362b7605f5c12d0531
|
|
| MD5 |
b5297882972e4e1f6a8c08ac912a1bc4
|
|
| BLAKE2b-256 |
6744d4718187190547845aab3cfb76fcca49823c051b66bb752a984ffa7984d5
|







