Predict ancestral sequence of fungal repeat elements by correcting for RIP-like mutations in multi-sequence DNA alignments.

Project description




██████╗ ███████╗██████╗ ██╗██████╗ ██████╗
██╔══██╗██╔════╝██╔══██╗██║██╔══██╗╚════██╗
██║ ██║█████╗ ██████╔╝██║██████╔╝ █████╔╝
██║ ██║██╔══╝ ██╔══██╗██║██╔═══╝ ██╔═══╝
██████╔╝███████╗██║ ██║██║██║ ███████╗
╚═════╝ ╚══════╝╚═╝ ╚═╝╚═╝╚═╝ ╚══════╝
deRIP2 scans aligned sequences for evidence of un-RIP'd precursor states, allowing for improved RIP-correction across large repeat families in which members are independently RIP'd.
Use deRIP2 to:
-
Predict ancestral fungal transposon sequences by correcting for RIP-like mutations (CpA --> TpA) and cytosine deamination (C --> T) events.
-
Mask RIP or deamination events as ambiguous bases to remove RIP signal from phylogenetic analyses.
Table of contents
Installation
Install from PyPi.
pip install derip2
Pip install latest development version from GitHub.
pip install git+https://github.com/Adamtaranto/deRIP2.git
Test installation.
# Print version number and exit.
derip2 --version
# Get usage information
derip2 --help
Setup Development Environment
If you want to contribute to the project or run the latest development version, you can clone the repository and install the package in editable mode.
# Clone repository
git clone https://github.com/Adamtaranto/deRIP2.git && cd deRIP2
# Create virtual environment
conda env create -f environment.yml
# Activate environment
conda activate derip2-dev
# Install package in editable mode
pip install -e '.[dev]'
Example usage
For aligned sequences in 'mintest.fa':
- Any column with >= 70% gap positions will not be corrected and a gap inserted in corrected sequence.
- Bases in column must be >= 80% C/T or G/A
- At least 50% bases in a column must be in RIP dinucleotide context (C/T as CpA / TpA) for correction.
- Default: Inherit all remaining uncorrected positions from the least RIP'd sequence.
- Mask all substrate and product motifs from corrected columns as ambiguous bases (i.e. CpA to TpA --> YpA)
Basic usage with masking
derip2 -i tests/data/mintest.fa \
--max-gaps 0.7 \
--max-snp-noise 0.2 \
--min-rip-like 0.5 \
--mask \
-d results \
--prefix derip_output
Output:
results/derip_output.fasta- Corrected sequenceresults/derip_output_alignment.fasta- Alignment with masked correctionsresults/derip_output_masked_alignment.fasta- Alignment with masked corrections
With vizualization
The --plot option will create a visualization of the alignment with RIP markup. The --plot-rip-type option can be used to specify the type of RIP events to be displayed in the alignment visualization product, substrate, or both.
derip2 -i tests/data/mintest.fa \
--max-gaps 0.7 \
--max-snp-noise 0.2 \
--min-rip-like 0.5 \
--plot \
--plot-rip-type both \
-d results \
--prefix derip_output
Output:
results/derip_output.fasta- Corrected sequenceresults/derip_output_masked_alignment.fasta- Alignment with masked correctionsresults/derip_output_visualization.png- Visualization of the alignment with RIP markup
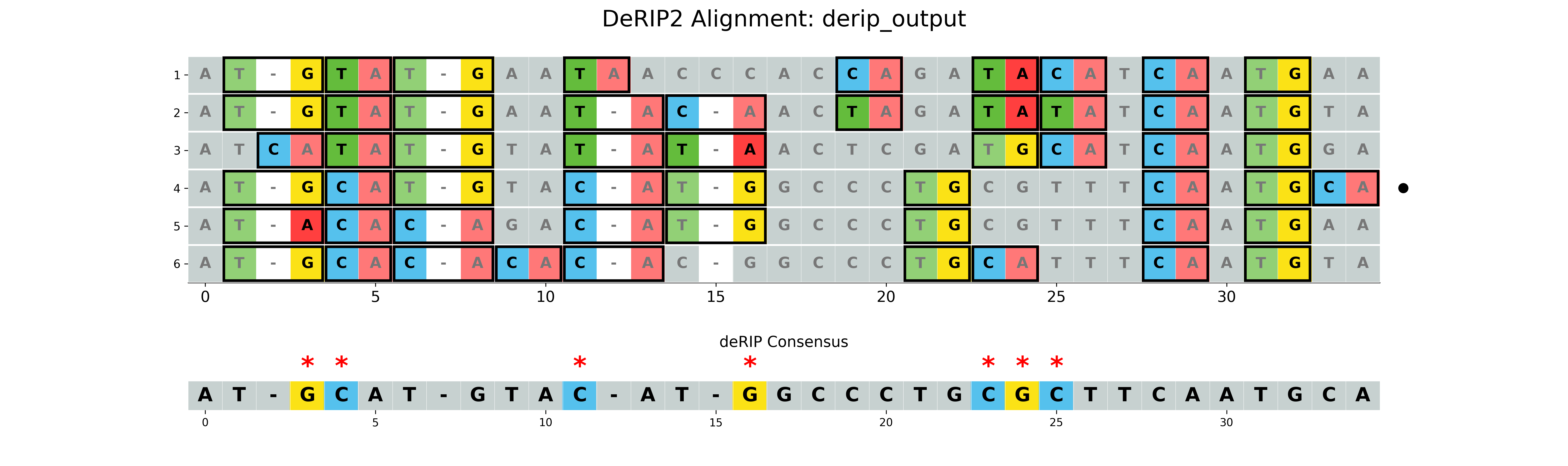
Using maximum GC content for filling
By default uncorrected positions in the output sequence are filled from the sequence with the lowest RIP count. If the --fill-max-gc option is set, remaining positions are filled from the sequence with the highest G/C content sequence instead.
derip2 -i tests/data/mintest.fa \
--max-gaps 0.7 \
--max-snp-noise 0.2 \
--min-rip-like 0.5 \
--fill-max-gc \
-d results \
--prefix derip_gc_filled
Alternatively, the --fill-index option can be used to force selection of alignment row to fill uncorrected positions from by row index number (indexed from 0). Note: This will override the --fill-max-gc option.
Correcting all deamination events
If the --reaminate option is set, all deamination events will be corrected, regardless of RIP context.
--plot-rip-type product is used to highlight the product of RIP events in the visualization.
Non-RIP deamination events are also highlighted.
derip2 -i tests/data/mintest.fa \
--max-gaps 0.7 \
--reaminate \
-d results \
--plot \
--plot-rip-type product \
--prefix derip_reaminated
Output:
results/derip_reaminated.fasta- Corrected sequence using highest GC content sequence for fillingresults/derip_reaminated_alignment.fasta- Alignment with corrected sequence appendedresults/derip_reaminated_vizualization.png- Visualization of the alignment with RIP markup
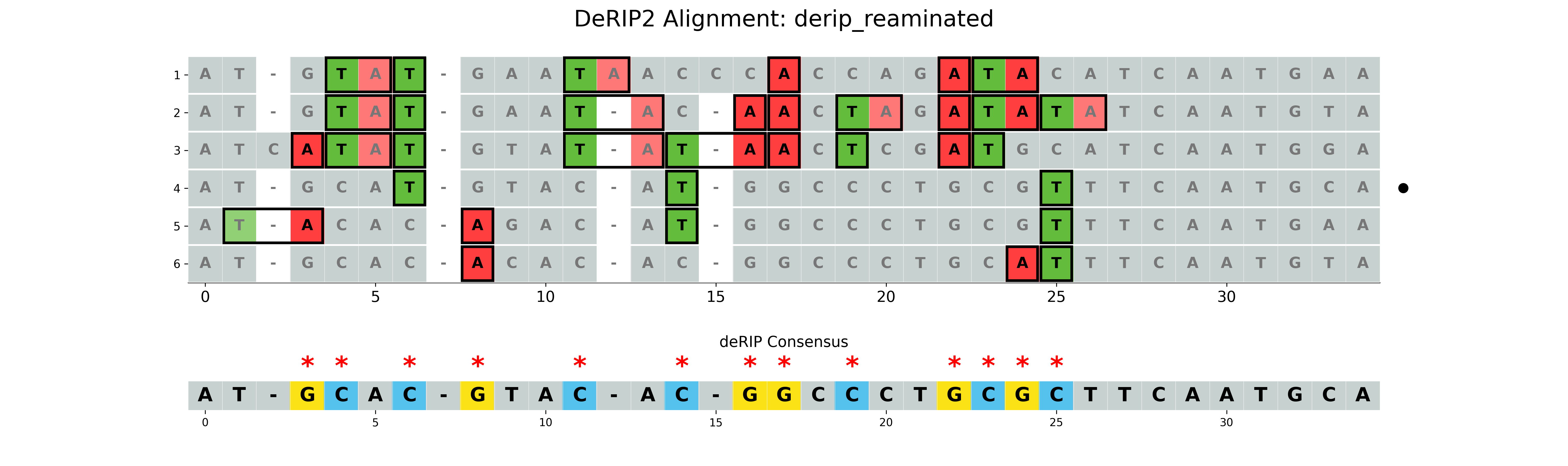
Standard Options
--version Show the version and exit.
-i, --input TEXT Multiple sequence alignment. [required]
-g, --max-gaps FLOAT Maximum proportion of gapped positions in
column to be tolerated before forcing a gap
in final deRIP sequence. [default: 0.7]
-a, --reaminate Correct all deamination events independent
of RIP context.
--max-snp-noise FLOAT Maximum proportion of conflicting SNPs
permitted before excluding column from
RIP/deamination assessment. i.e. By default
a column with >= 0.5 'C/T' bases will have
'TpA' positions logged as RIP events.
[default: 0.5]
--min-rip-like FLOAT Minimum proportion of deamination events in
RIP context (5' CpA 3' --> 5' TpA 3')
required for column to deRIP'd in final
sequence. Note: If 'reaminate' option is set
all deamination events will be corrected.
[default: 0.1]
--fill-max-gc By default uncorrected positions in the
output sequence are filled from the sequence
with the lowest RIP count. If this option is
set remaining positions are filled from the
sequence with the highest G/C content.
--fill-index INTEGER Force selection of alignment row to fill
uncorrected positions from by row index
number (indexed from 0). Note: Will override
'--fill-max-gc' option.
--mask Mask corrected positions in alignment with
degenerate IUPAC codes.
--no-append If set, do not append deRIP'd sequence to
output alignment.
-d, --out-dir TEXT Directory for deRIP'd sequence files to be
written to.
-p, --prefix TEXT Prefix for output files. Output files will
be named prefix.fasta,
prefix_alignment.fasta, etc. [default:
deRIPseq]
--plot Create a visualization of the alignment with
RIP markup.
--plot-rip-type [both|product|substrate]
Specify the type of RIP events to be
displayed in the alignment visualization.
[default: both]
--loglevel [DEBUG|INFO|WARNING|ERROR|CRITICAL]
Set logging level. [default: INFO]
--logfile TEXT Log file path.
-h, --help Show this message and exit.
Algorithm overview
For each column in input alignment:
- Check if number of gapped rows is greater than max gap proportion. If true, then a gap is added to the output sequence.
- Set invariant column values in output sequence.
- If at least X proportion of bases are C/T or G/A (i.e.
max-snp-noise= 0.4, then at least 0.6 of positions in column must be C/T or G/A). - If reaminate option is set then revert T-->C or A-->G.
- If reaminate is not set then check for number of positions in RIP dinucleotide context (C/TpA or TpG/A).
- If proportion of positions in column in RIP-like context =>
min-rip-likethreshold, AND at least one substrate and one product motif (i.e. CpA and TpA) is present, perform RIP correction in output sequence. - For all remaining positions in output sequence (not filled by gap, reaminate, or RIP-correction) inherit sequence from input sequence with the fewest observed RIP events (or greatest GC content if RIP is not detected or multiple sequences sharing min-RIP count).
Issues
Submit feedback to the Issue Tracker
License
Software provided under MIT license.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file derip2-0.4.1.tar.gz.
File metadata
- Download URL: derip2-0.4.1.tar.gz
- Upload date:
- Size: 590.3 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: python-httpx/0.28.1
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
1f11636b3abb95ead6dba5936dcb920c83b55073c8217f6599ffa8ef7006e78a
|
|
| MD5 |
dd0617c89556e0dd5672f7367c7eec32
|
|
| BLAKE2b-256 |
9e727a51446e64ea866a833e74647d68df1dcd2844c39ee60fe63df16515b4c5
|
File details
Details for the file derip2-0.4.1-py3-none-any.whl.
File metadata
- Download URL: derip2-0.4.1-py3-none-any.whl
- Upload date:
- Size: 46.7 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: python-httpx/0.28.1
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
04c183c2de51635a3a99469df3290a42b7ea59b406b1703d8b155b0921257e4c
|
|
| MD5 |
99390ff0d3aaa44b5f7cdfbb2a9af720
|
|
| BLAKE2b-256 |
c4779c3e67f28da52769447bd740b0c122eadb2764c28babfefd8a029c81cad2
|







