Fast and scalable phylogenetic tree viewer for multi-omic data analysis


Project description
Empress



Introduction
Empress is a fast and scalable phylogenetic tree viewer.
Empress helps users explore the hierarchical relationships between features in a dataset. Any type of "feature" can be viewed in this way: historically these features have often represented evolutionary relationships of species in community surveys, but you can view pretty much any type of information with hierarchical organization. For example, we could view trees of amplicon sequence variants (ASVs) or operational taxonomic units (OTUs) generated from 16S rRNA marker gene sequencing data, trees generated from shotgun metagenomics sequencing data, or trees of metabolomics data generated using Qemistree (just to name a few options).
Empress supports categorically new functionality, such as integration and synchronized animations with ordination plots, as well as functionality common to established tree viewers (e.g. metadata coloring, clade collapsing, and barplots).
Screenshot
Installation & Basic Usage
Empress is available as either a standalone program or a QIIME 2 plugin. The standalone version will generate a folder with the HTML/JS/CSS files necessary to view the plot while the QIIME 2 version will generate a .qzv Visualization that can be viewed on https://view.qiime2.org/ or by using qiime tools view.
Standalone Version
Empress is available through PyPI. We recommend installing Empress into an environment (e.g. a conda environment) using a Python version of at least 3.6.
Run the following commands to install Empress:
pip install cython "numpy >= 1.12.0"
pip install empress
Try running the command empress --help to ensure that Empress has been installed properly. If you see details for the different Empress commands then the installation has succeeded and you are ready to start using Empress!
Available commands
Empress provides two commands: empress tree-plot and empress community-plot. Both commands generate an Empress visualization, but community-plot requires you to pass in a feature table and sample metadata while tree-plot only requires a tree file. See this section of the docs for some more details.
Input files
The standalone version of Empress takes the following filetypes as inputs. (Note that for empress tree-plot all of these except for the tree are optional, and for empress community-plot all except for the tree, feature table, and sample metadata are optional.)
| Input | Filetype |
|---|---|
| Tree | Newick |
| Feature Table | BIOM |
| Sample Metadata | TSV |
| Feature Metadata | TSV |
| PCoA | scikit-bio OrdinationResults |
Example standalone usage
empress tree-plot
# Option 1: Using "long" parameter names
empress tree-plot \
--tree tree.nwk \
--feature-metadata feature-metadata.tsv \
--output-dir tree-viz
# Option 2: Using "short" parameter names
empress tree-plot -t tree.nwk -fm feature-metadata.tsv -o tree-viz
empress community-plot
# Option 1: Using "long" parameter names
empress community-plot \
--tree tree.nwk \
--table feature-table.biom \
--sample-metadata sample-metadata.tsv \
--feature-metadata feature-metadata.tsv \
--pcoa ordination.txt \
--filter-extra-samples \
--output-dir community-tree-viz
# Option 2: Using "short" parameter names
empress community-plot \
-t tree.nwk \
-tbl feature-table.biom \
-sm sample-metadata.tsv \
-fm feature-metadata.tsv \
-p ordination.txt \
--filter-extra-samples \
-o community-tree-viz
You can view the details of the command line arguments with empress tree-plot --help and empress community-plot --help. Note that the path provided to -o/--output-dir must not exist, as it will be created by Empress upon successful execution of the command. It is also worth noting that the standalone version of the Empress commands does not support providing multiple sample/feature metadata files. If you have, for example, multiple feature metadata files, you should combine them all into one file that you pass to Empress.
The output will be a directory containing an empress.html file and a support_files directory containing the JS/CSS files required to view the plot in your browser. If you provided a PCoA to the community-plot command there will also be an emperor-resources subdirectory containing the files required to view the Emperor plot alongside the tree. You can view the empress.html file in any modern browser to interact with it the same way you would the QIIME 2 Visualization.
QIIME 2 Version
See the QIIME 2 installation page for instructions on how to install QIIME 2. Once you have QIIME 2 installed, make sure the conda environment is activated by running:
conda activate qiime2-2020.8
You can replace qiime2-2020.8 above with whichever version of QIIME 2 you have currently installed.
Now, run the following command to install Empress using PyPI:
pip install empress
Once you have installed Empress, run the following commands to ensure that Empress was installed correctly. If you see information about Empress' QIIME 2 plugin, the installation was successful!
qiime dev refresh-cache
qiime empress --help
Example QIIME 2 usage
qiime empress tree-plot
qiime empress tree-plot \
--i-tree tree.qza \
--m-feature-metadata-file taxonomy.qza \
--o-visualization tree-viz.qzv
qiime empress community-plot
qiime empress community-plot \
--i-tree tree.qza \
--i-feature-table feature-table.qza \
--m-sample-metadata-file sample-metadata.tsv \
--m-feature-metadata-file taxonomy.qza \
--i-pcoa ordination.qza \
--p-filter-extra-samples \
--o-visualization community-tree-viz.qzv
Tutorial: Using Empress in QIIME 2
In this tutorial, we'll use Empress through QIIME 2 and demonstrate its basic usage with the Moving Pictures tutorial dataset. This dataset contains human microbiome samples from two individuals at four body sites across five timepoints.
First, a note about Empress' commands
Empress currently has two commands available:
$ qiime empress --help
Usage: qiime empress [OPTIONS] COMMAND [ARGS]...
Description: This QIIME 2 plugin wraps Empress and supports interactive
visualization of phylogenetic trees.
Plugin website: http://github.com/biocore/empress
Getting user support: Please post to the QIIME 2 forum for help with this
plugin: https://forum.qiime2.org
Options:
--version Show the version and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
community-plot Visualize phylogenies and community data with Empress (and,
optionally, Emperor)
tree-plot Visualize phylogenies with Empress
Both of these commands generate similar visualizations. The functionality available in a visualization created by qiime empress community-plot is a superset of the functionality available in a visualization created by qiime empress tree-plot: tree-plot is useful if you don't have a table and just want to visualize a tree (optionally with feature metadata). Here, we're going to be using community-plot, but much of this tutorial is also applicable to tree-plot.
Downloading Input Artifacts and Metadata
Before we start, we’ll need to download the necessary input artifacts for running qiime empress community-plot. The first four of these artifacts are produced during the Moving Pictures tutorial, and the last artifact was produced afterwards using data from the tutorial. These artifacts are:
- A feature table (a QIIME 2 artifact of type
FeatureTable[Frequency]) - A sample metadata file (a tab-separated-value file)
- A rooted tree (a QIIME 2 artifact of type
Phylogeny[Rooted]) - Taxonomic assignments of our features (a QIIME 2 artifact of type
FeatureData[Taxonomy]) - A PCoA biplot results file (a QIIME 2 artifact of type
PCoAResults % Properties('biplot'))- This artifact in particular was produced by the
qiime diversity pcoaplugin, but ordinations / biplots created by other tools (e.g. DEICODE) also work well with Empress.
- This artifact in particular was produced by the
The last item is required only when displaying an Empress tree plot in tandem with an Emperor PCoA plot/biplot (a.k.a. an Empire plot!)
You can download these files individually by clicking the links below or by using wget to download them directly from your terminal.
table.qzaview | downloadsample_metadata.tsvdownloadrooted-tree.qzaview | downloadtaxonomy.qzaview | downloadbiplot.qzaview | download
First we’ll create a directory which we'll download our files to and move into it:
mkdir empress-tutorial
cd empress-tutorial
Now we'll download the files using wget:
wget https://docs.qiime2.org/2019.10/data/tutorials/moving-pictures/table.qza
wget https://data.qiime2.org/2019.10/tutorials/moving-pictures/sample_metadata.tsv
wget https://docs.qiime2.org/2019.10/data/tutorials/moving-pictures/rooted-tree.qza
wget https://docs.qiime2.org/2019.10/data/tutorials/moving-pictures/taxonomy.qza
wget https://raw.githubusercontent.com/biocore/empress/master/docs/moving-pictures/biplot.qza
We are now ready to visualize this data using Empress.
Empress Plot
We’ll start by creating a simple stand-alone tree visualization artifact, which will enable us to explore the tree and associated data using the various functionalities available in Empress.
qiime empress community-plot \
--i-tree rooted-tree.qza \
--i-feature-table table.qza \
--m-sample-metadata-file sample_metadata.tsv \
--m-feature-metadata-file taxonomy.qza \
--o-visualization empress-tree.qzv
To view the newly made empress-tree.qzv artifact, you can drag and drop the file onto https://view.qiime2.org/ or load it locally by running:
qiime tools view empress-tree.qzv
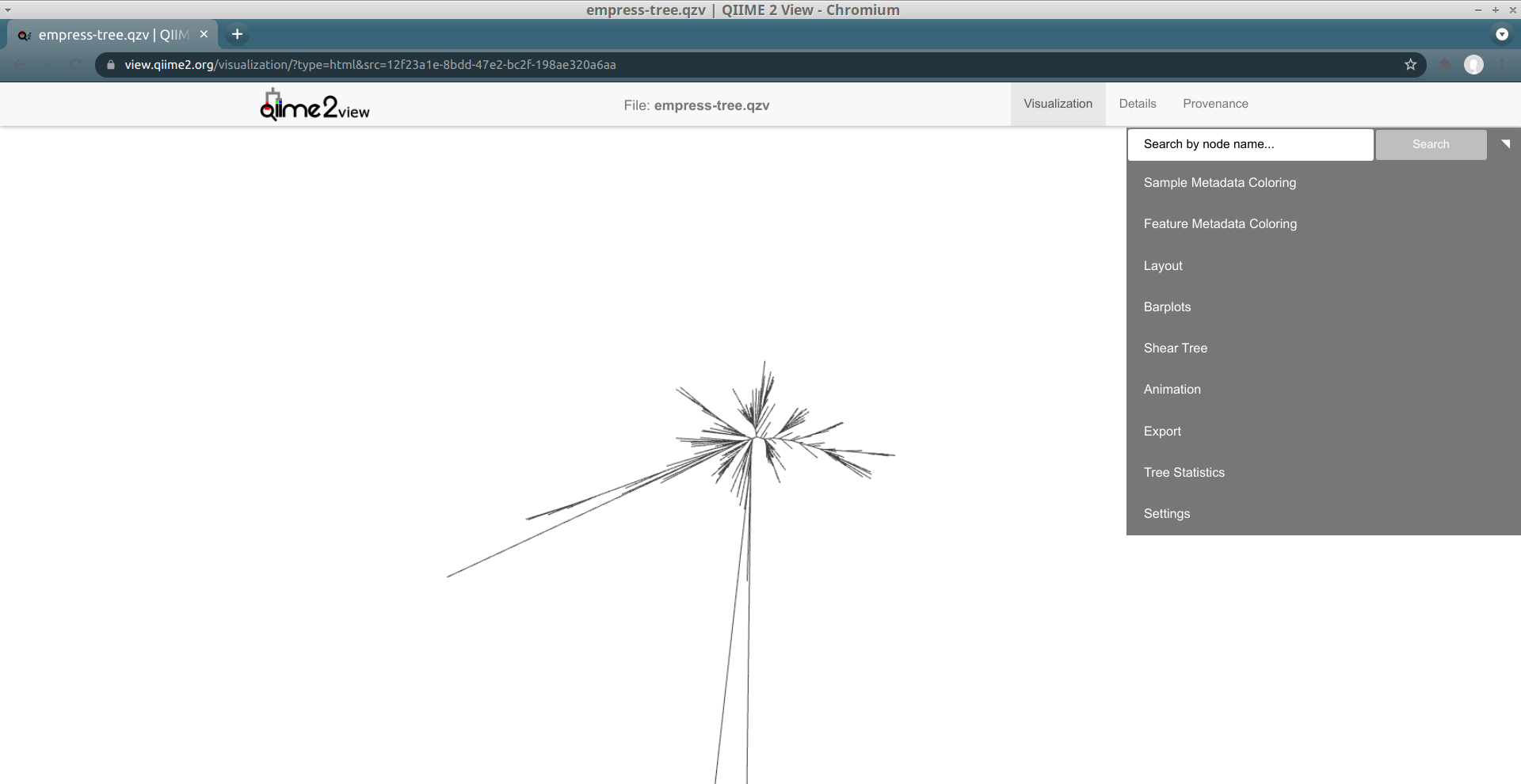
The starting plot is a simple unrooted tree which has all the normal properties of a phylogenetic tree. The outermost “tips” of the tree are also referred to as “leaves”, “terminal nodes”, or “external nodes” and here represent a unique ASV. The line connected to a tip is referred to as a “branch”. A branch connects two or more nodes, or in this case a tip to an internal node. These internal nodes represent a divergent point between nodes and the branch length represents the evolutionary distance between divergence points. You can use your mouse’s scroll wheel to zoom in and out, and click and drag anywhere on the plot to move the display to take a closer look at the various tree components. On the top-right we see a display menu with several subcategories that allow us to customize the plot. We will explore these options in more detail below.
Exploring individual features
The first thing you likely noticed in this plot is the presence of several very long branches that stand out relative to the others. Let’s investigate these further. Using your computer mouse, move the display to focus in on the tip of the longest branch and click on the node.
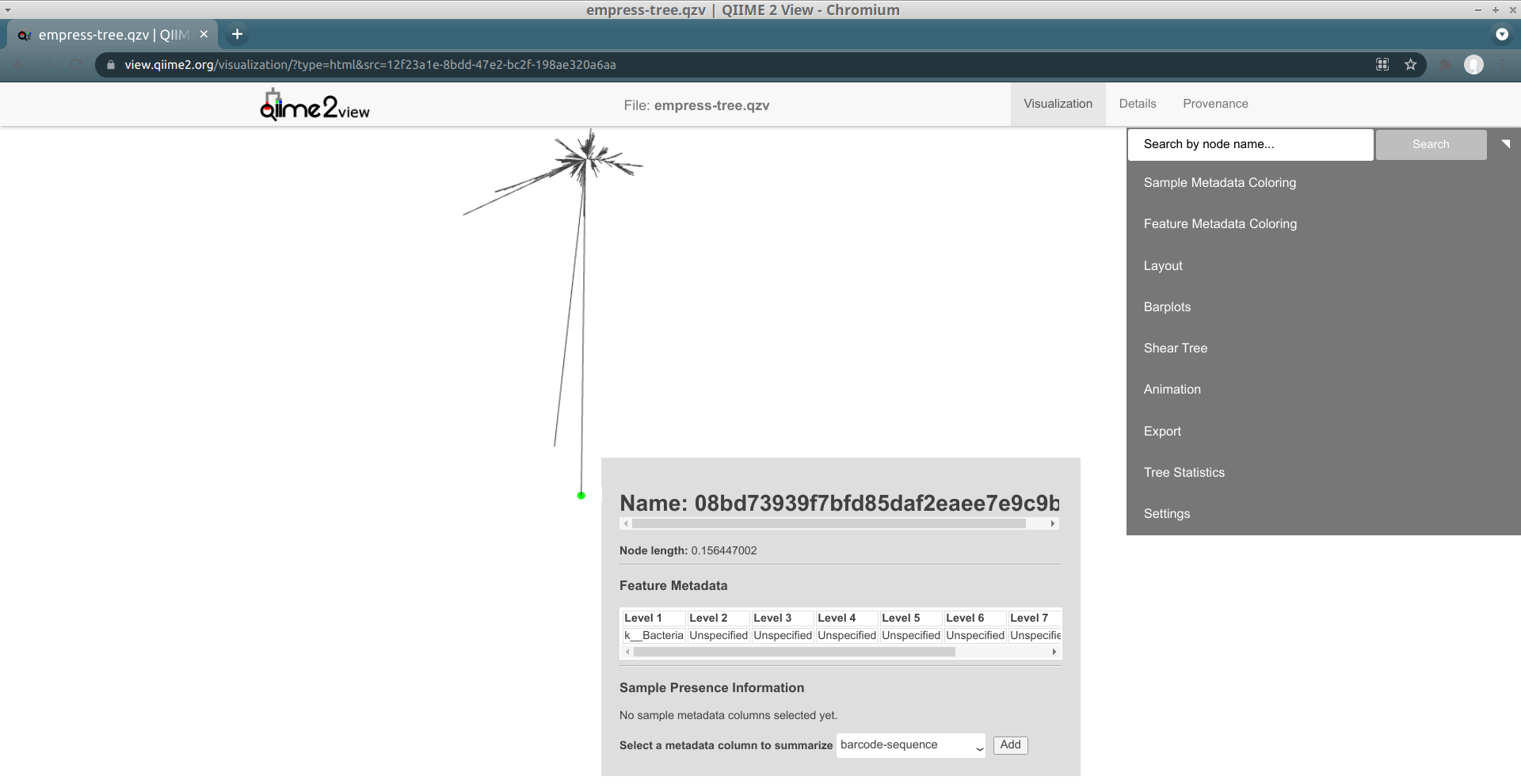
A new menu appears with details about the selected node, including its name and taxonomic assignment. You’ll notice that this feature has only been classified at the Kingdom level, meaning that our feature-classifier was not able to find a suitable match in the reference database used to assign these taxonomy classifications (in this case, Greengenes). More often than not, these features correspond to non-biological reads such as chimeras, contaminants, or reads that have index-hopped from other samples. We will explore these possibilities further later.
We should also note that the tree used in this tutorial was built using the common de novo tree-building approach, and it has previously been shown that the presence of these outlier branches in de novo trees can lead to artificial clustering of samples (Janssen et. al 2018).
In this window we can also view details about sample metadata related to this feature. From the drop-down menu select body-site and click the Add button. A new Sample Presence Information summary table appears which displays the number of samples containing the selected feature.
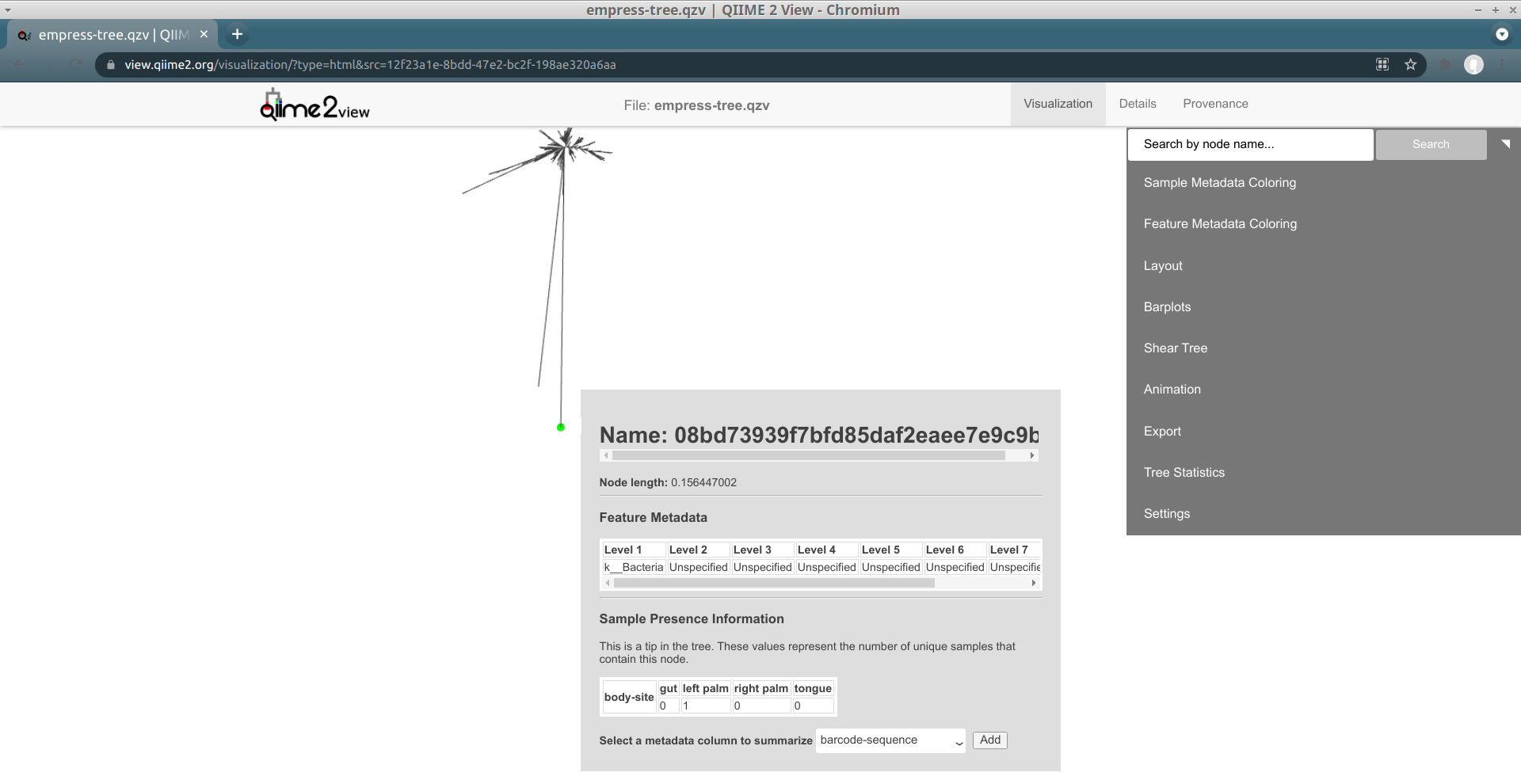
We can see that our ASV is present in only 1 left palm sample. You can select multiple metadata columns. While the table here does not give us information about the abundance of this feature, we can easily search the feature name in the feature table summary visualization artifact created previously in the QIIME 2 Moving Pictures tutorial. From there we see that this particular feature has a total abundance of 2, which is another strong indicator of a non-biological read. Try clicking the tips in a few other outlier branches. Do you see a similar pattern? Now try clicking on a tip of one of the shorter branches. Notice the much improved classification!
We can also locate specific features of interest using the search bar at the top of the main menu. For example, in our feature-table the most abundant ASV is 4b5eeb300368260019c1fbc7a3c718fc. Paste this name in to the search bar and click Search.
This feature’s tip in the tree is now highlighted with a bright green circle.
It looks like this ASV is a species belonging to the Bacteroides genus, and
is present in all of the four "body sites" included in this data (although it's
only present in one tongue sample).
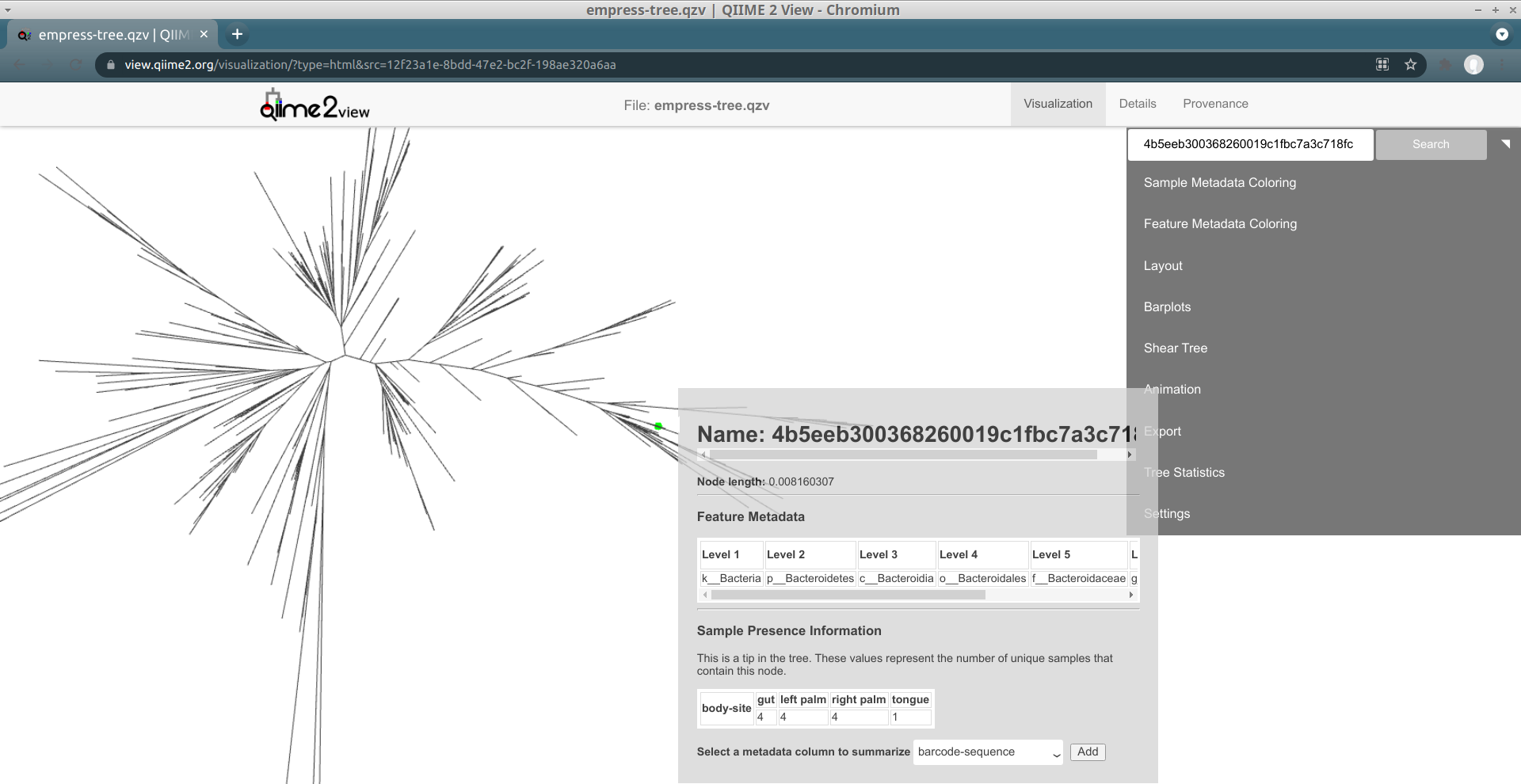
Exploring groups of features
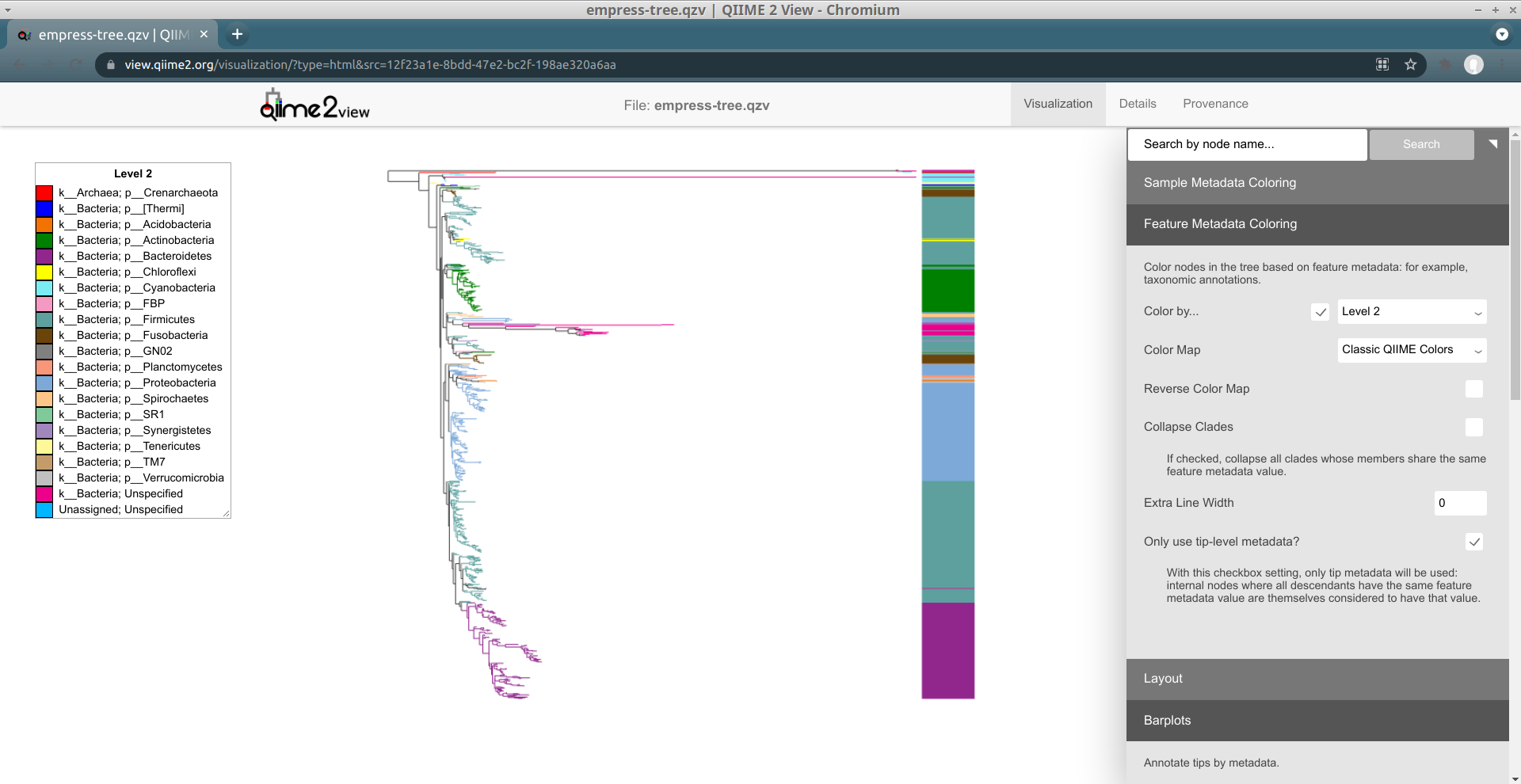
Another way of exploring the classification of our features is to color the branches based on their taxonomic designation. From the main menu, click Feature Metadata Coloring, check the Color by… box, select Level 2 (which here corresponds to the phylum level), and click Update.
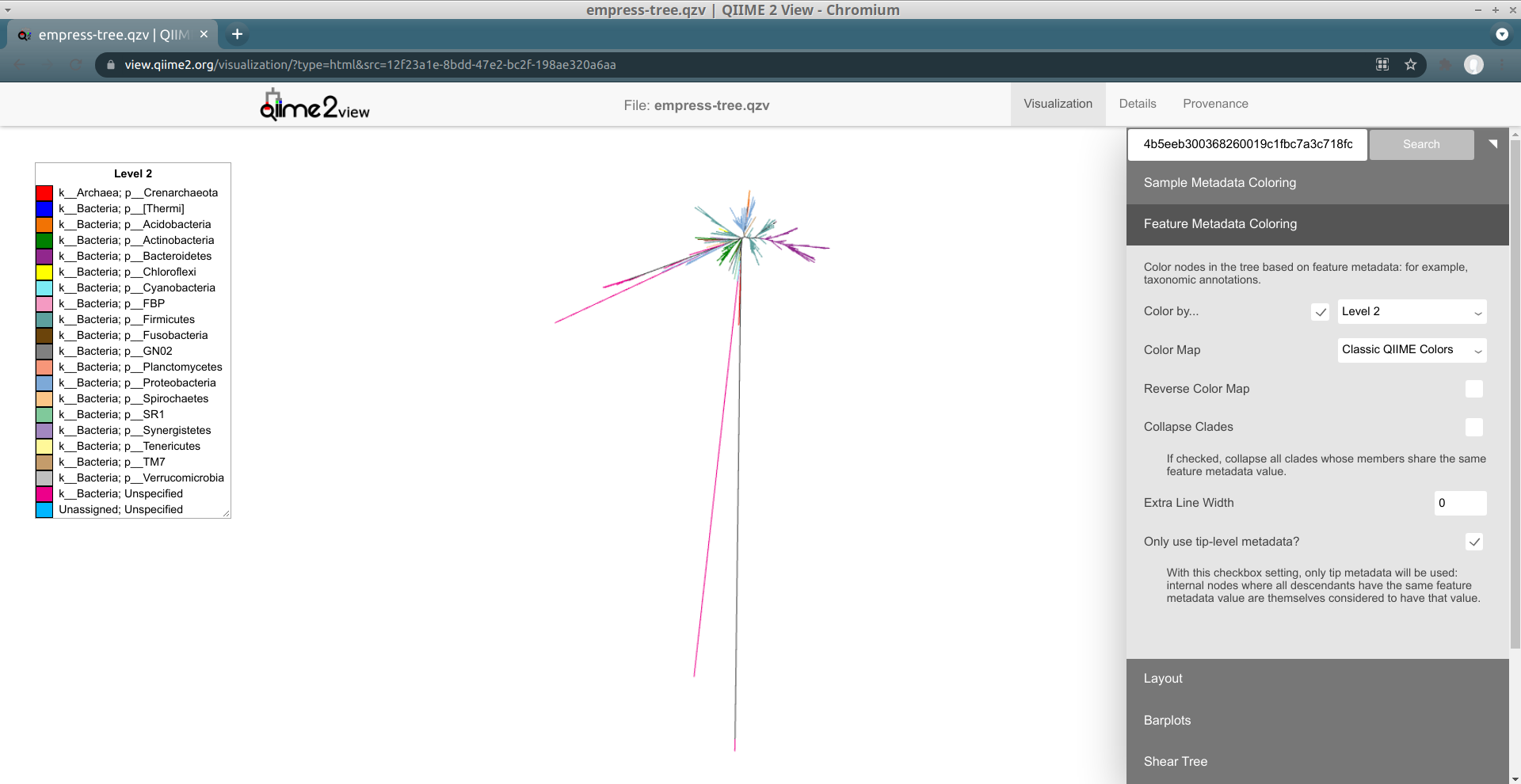
The plot is now updated so each branch is now colored by its phylum-level classification. We can see that many of the extra long branches are now mostly the same magenta color. Check out the legend on the left side of the screen -- it turns out that the magenta color corresponds to a phylum-level classification of k__Bacteria; Unspecified, indicating that these ASVs were only classified as Bacteria. You may also have noticed that these outlier branches appear mainly in 2 distinct clusters. While we don’t have any more information about the classification of these features, perhaps we can gain some more insight regarding their classification by looking at their closest common ancestors that do have taxonomic information.
Exploring a feature’s closest common ancestors
So far, we’ve looked at our data using the default unrooted tree view. To visually locate these features’ closest common ancestors, it may be easier to switch to a different layout. From the main menu, click Layout then select Circular (or Rectangular). Our plot automatically switches to a rooted layout.
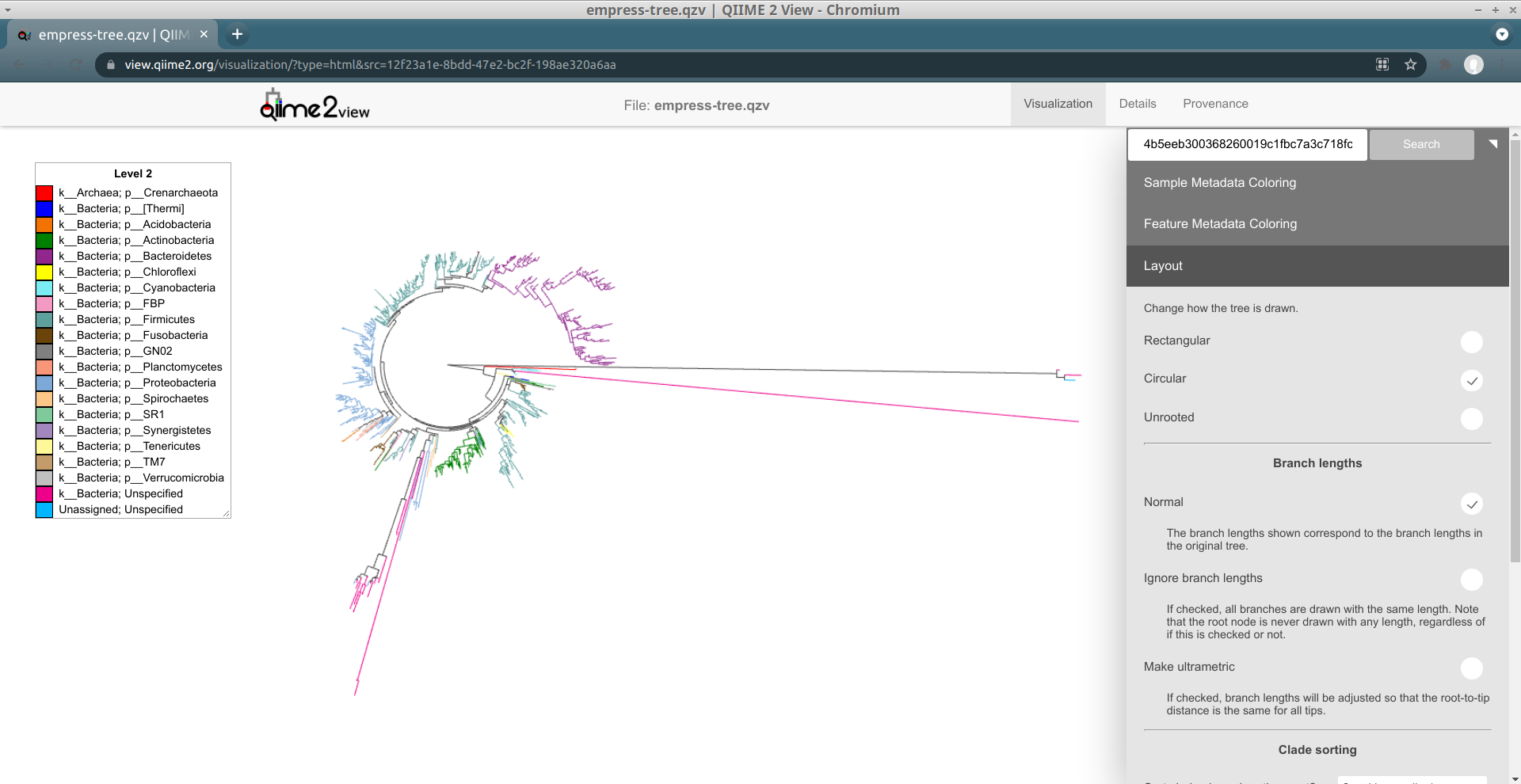
Now, let's zoom into the longest branch of the bottom cluster of k__Bacteria; Unspecified nodes and click on one of the close tips that has a different phylum classification (light blue).
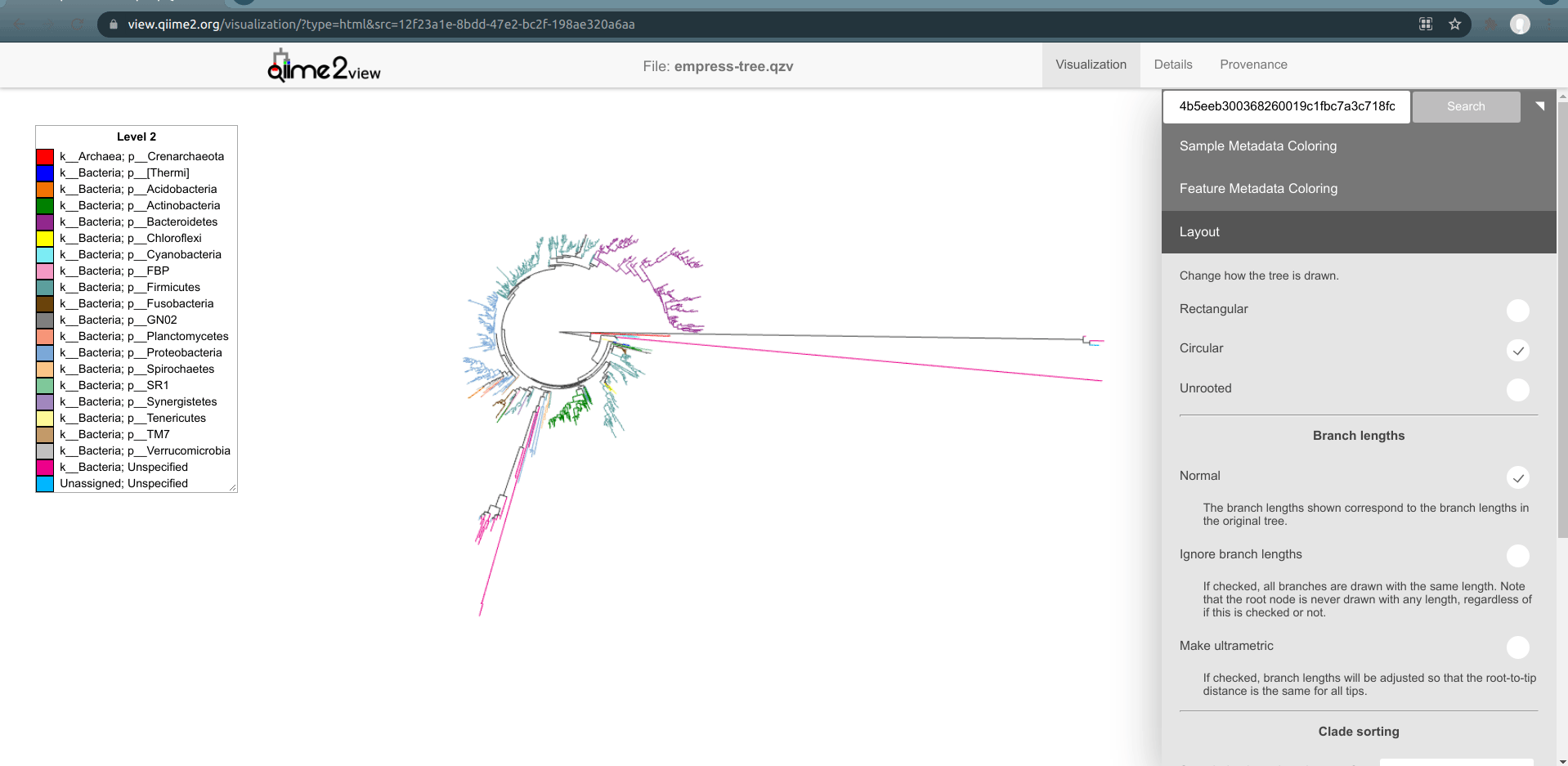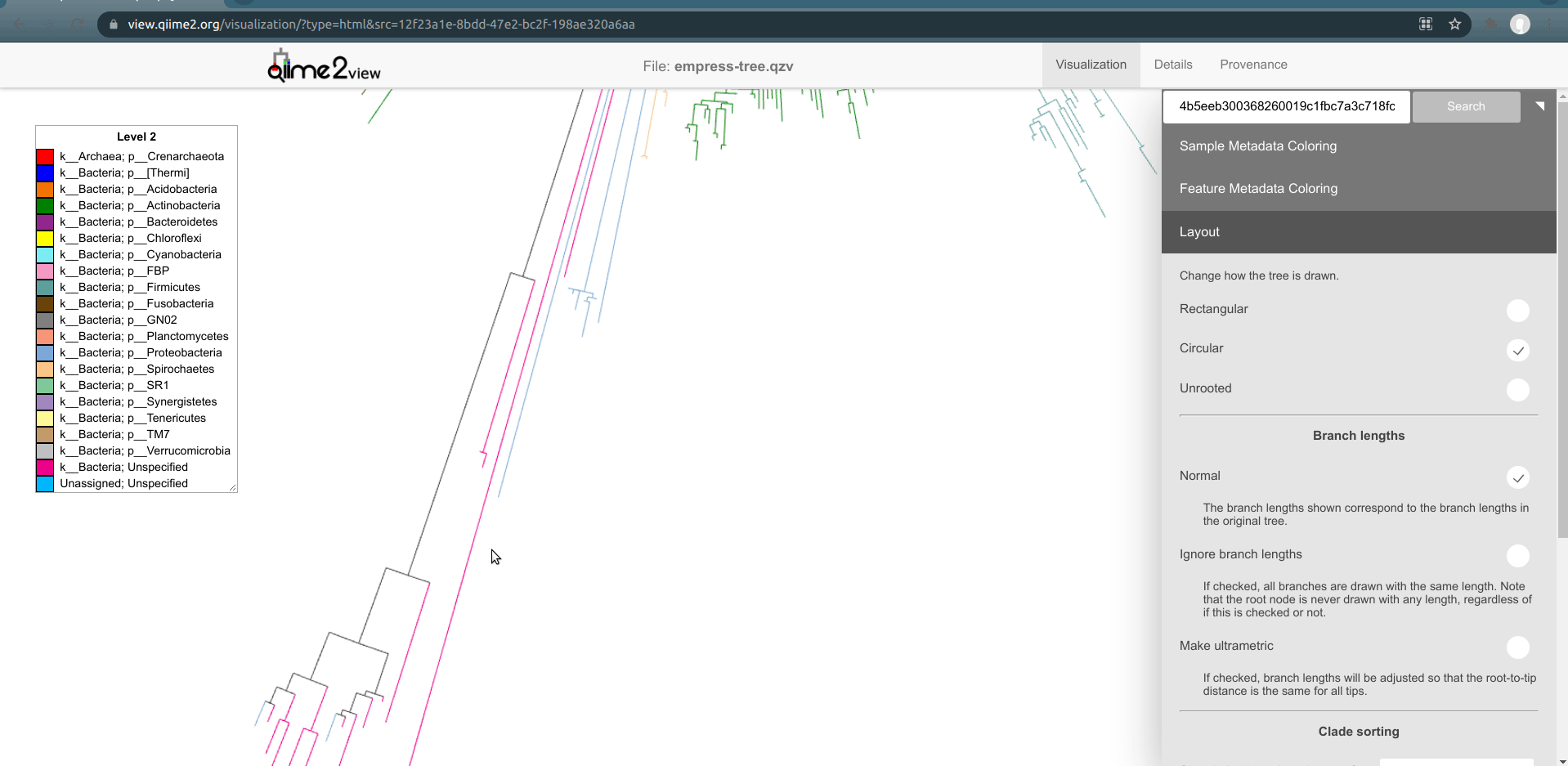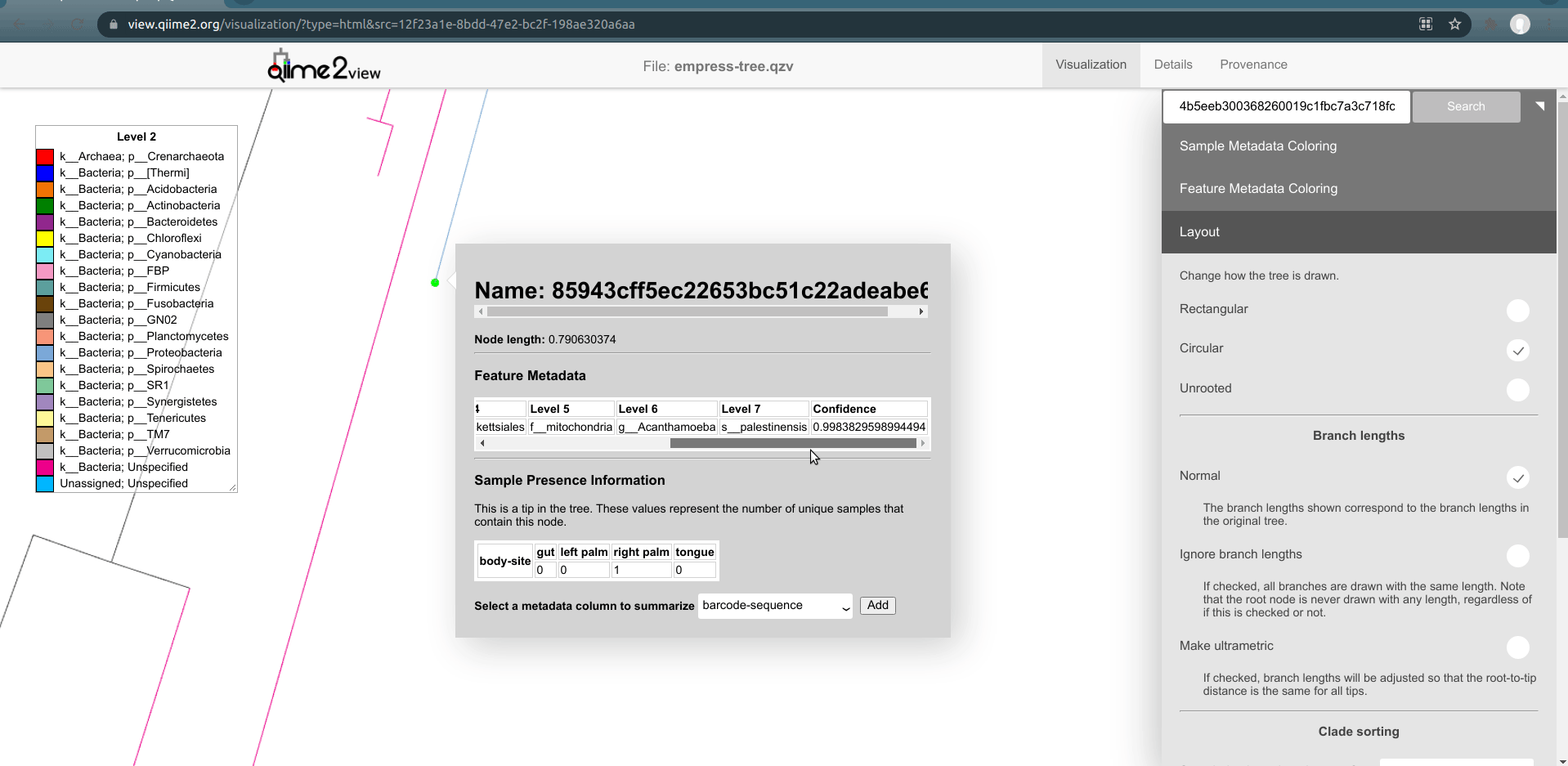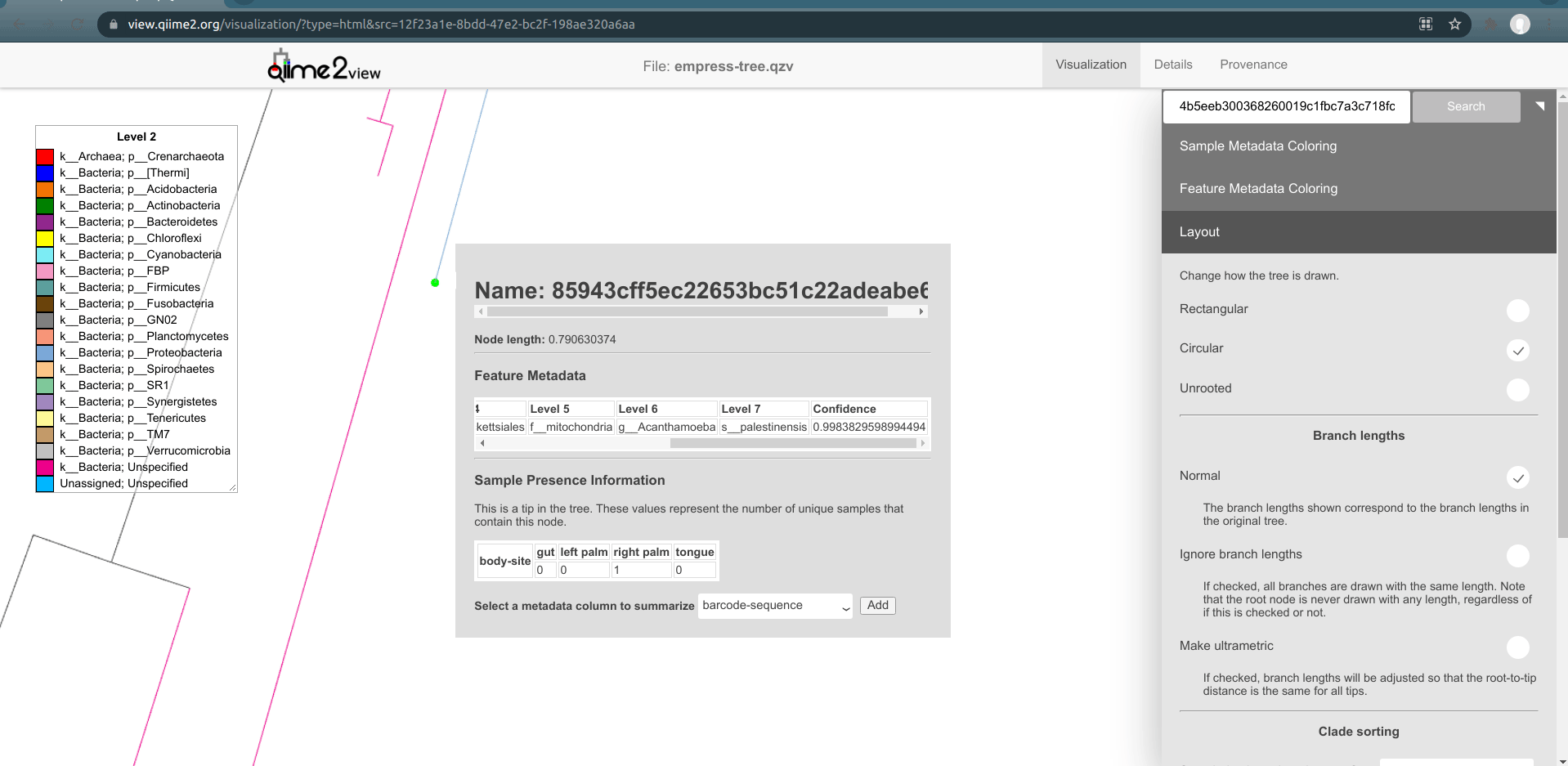
Interestingly, we see that this node is classified as Acanthamoeba Palestinensis which is actually not a bacteria but rather a protozoa. It is not uncommon for certain Eukaryotes to appear in bacterial/archaeal reference databases as they may share a similar genetic lineage. Remember that mitochondria and chloroplasts likely evolved from prokaryotes themselves. Explore a few other common ancestral nodes from different outlier branches. We can see other surprising appearances by Cucurbita pepo (a variety of squash or pumpkin), Raphanus sativus (radish), and Streptophyta (an order of plants). Based on these results one might speculate that our Unspecified features likely also belong to either plants or protozoa groups rather than bacteria. Further, since these features appear only on the palm samples, it’s possible the source of these are in fact environmental contaminants rather than common human microbes.
Summarizing things for these Unspecified-phylum features: in general, given their relatively long branch lengths, their presence in few samples in the study in some cases at relatively low abundance, their lack of close matches in the reference database, and the fact that they are putatively related to non-microbial features, it may be safe to filter them from our table as non-biologically relevant reads. (That conclusion is just based on the results of this exploratory analysis, not a strict guideline.)
Identifying group-specific features
The composition of microbial communities of the gut, tongue, and palms are very different from each other. Suppose we are interested in identifying which features are unique to each body-site and their evolutionary relationships. We can do this in Empress by colorizing our tree based on columns from our sample metadata file. From the main menu, click Sample Metadata Coloring, check the Color by… box, and from the drop-down menu select body-site. Click the Update button.
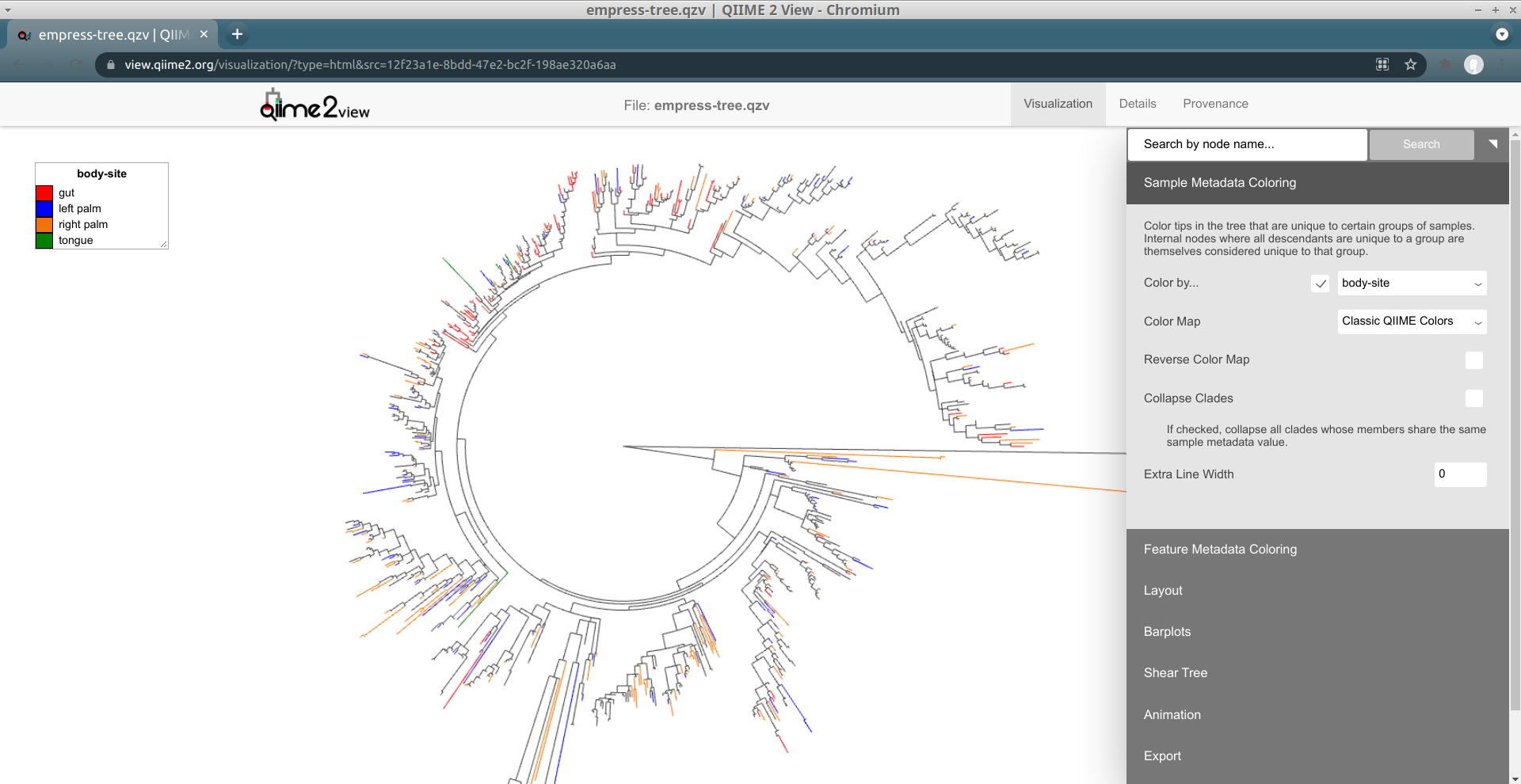
In this plot the colored branches represent lineages that are unique to the corresponding body site, while the uncolored branches are those that are shared across at least 2 body sites and thus cannot be displayed with a single color. While it is not surprising to see a large number of unique features in the gut samples (red) compared to the palm samples (blue and orange), it is interesting to see a large number of unique features between the left and right palm. Can you think of any biological reasons why the left and right palms may contain such different unique microbes? Even though the left and right palm do harbor unique features, the representative clades appear more integrated among themselves, suggesting that their phylogenies are still more similar to each other than the gut taxa which appear to cluster mainly among themselves.
Visualizing feature / sample metadata in barplots
Similarly to other tree visualization tools like iTOL, Empress can draw barplots in order to annotate tips of the tree with various types of information. Barplots are useful for doing this (moreso than node coloring, sometimes) because multiple "layers" of barplots can be shown at the same time -- this allows for us to view multiple types of data for the same tip simultaneously. Check out Figure 1 of Song and Sanders et al. 2020 for just one example of a tree visualization using multiple layers of barplots for a pretty and effective figure.
First: a small warning about barplots
Although barplots are very useful for identifying patterns, be wary of reading too much into them! The way the rectangular and circular layouts work means that a tip that looks "next" to another tip may actually be somewhat far away from that tip (e.g. in the rectangular layout if one tip is at the top of its clade, and another tip just "above" it is at the bottom of its clade). An example of this is shown below with the mustard and lavender clades:
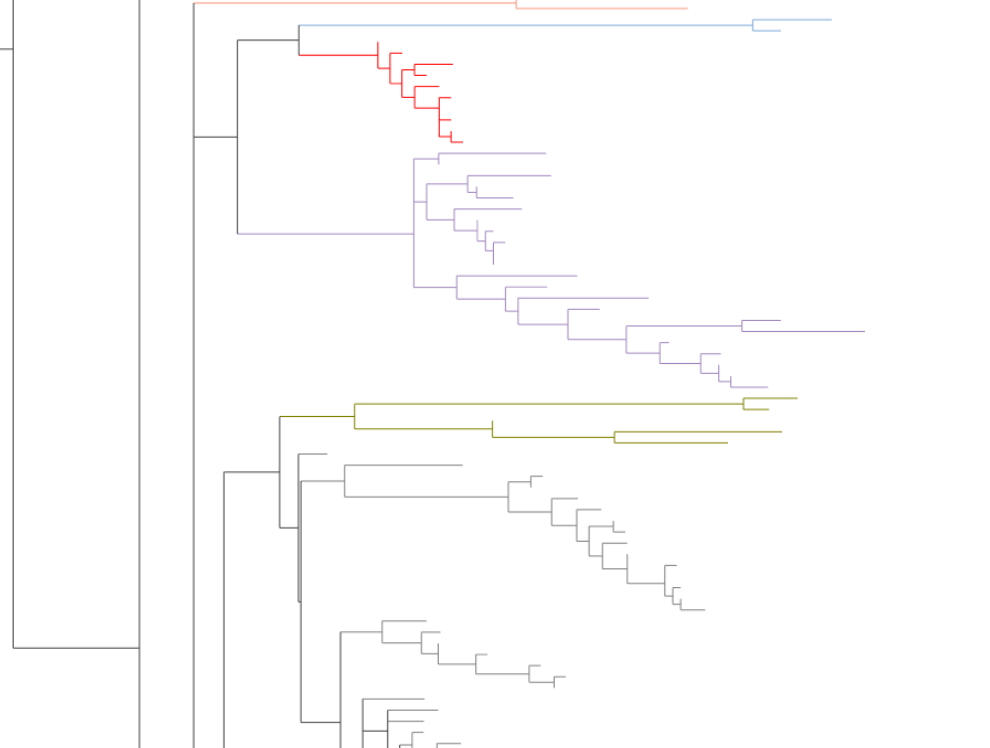
This can impact the way barplots look in ways that might not be immediately obvious. To quote "Inferring Phylogenies" (Felsenstein 2004), pages 573–574:
It is worth noting that by reordering tips, you can change the viewer's impression of the closeness of relationships. [...] A little judicious flipping may create a Great Chain of Being marching nicely along the sequence of names, even though the tree supports no such thing.
Diving into barplots: categorical feature metadata
Barplots in Empress are compatible with either the rectangular or circular layouts. Here we'll use the rectangular layout, but feel free to follow along with the circular layout if you prefer!
First off, change the layout to Rectangular (using the Layout section of the main menu), and then open up the Barplots section of the main menu and check the Draw Barplots? checkbox. Click the Update button that appears. By default, a red bar of uniform length will be drawn for every tip in the tree:
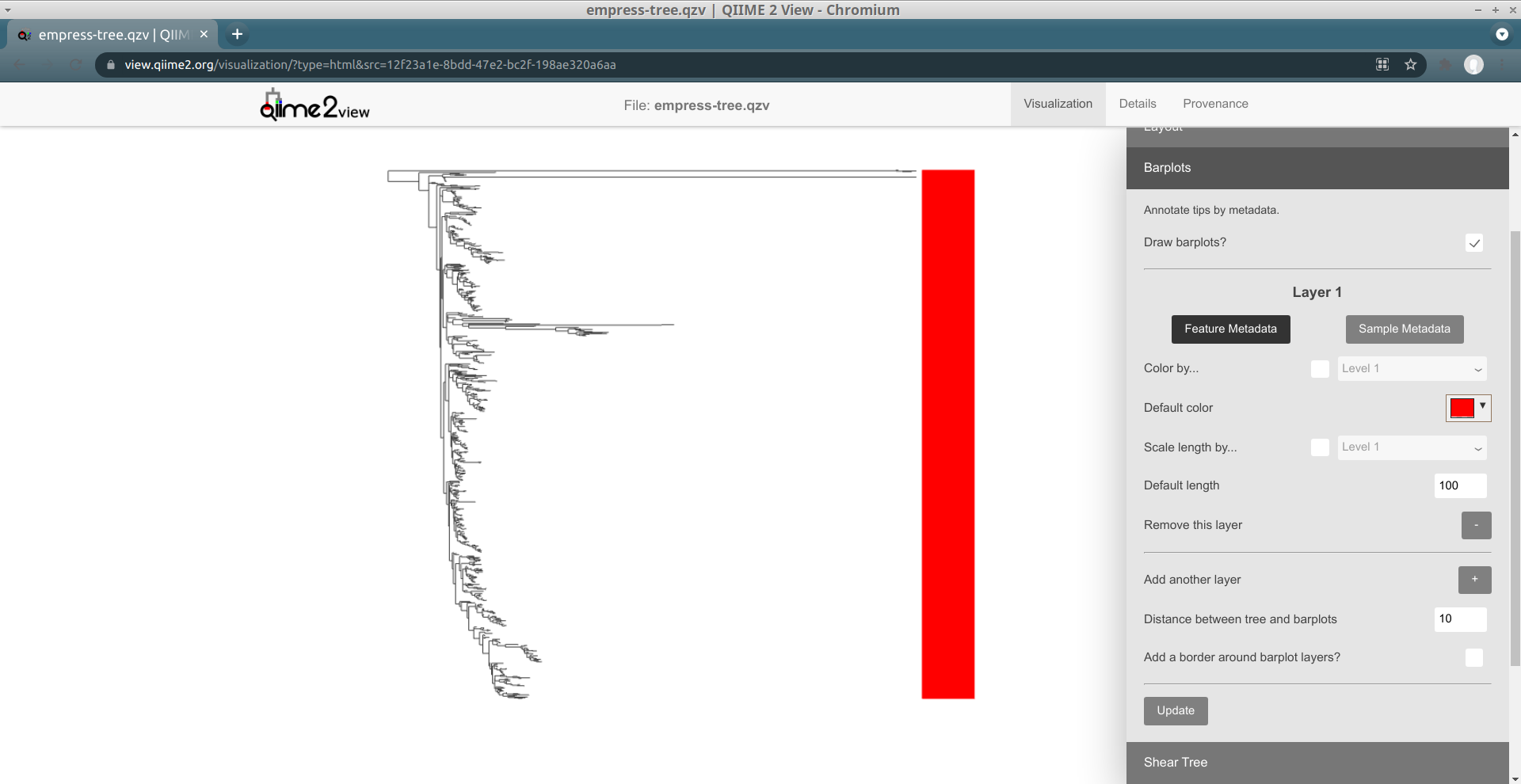
Although these bars are not very useful by default, we can encode them with information based on the feature or sample metadata you passed in to Empress when generating a visualization. Let's try coloring each tip's bar by its Level 2 feature metadata field (a.k.a. the phylum-level taxonomic assignments for the tips in this dataset): under the Layer 1 header, check the Color by... box, and from the drop-down menu select Level 2. Click the Update button.
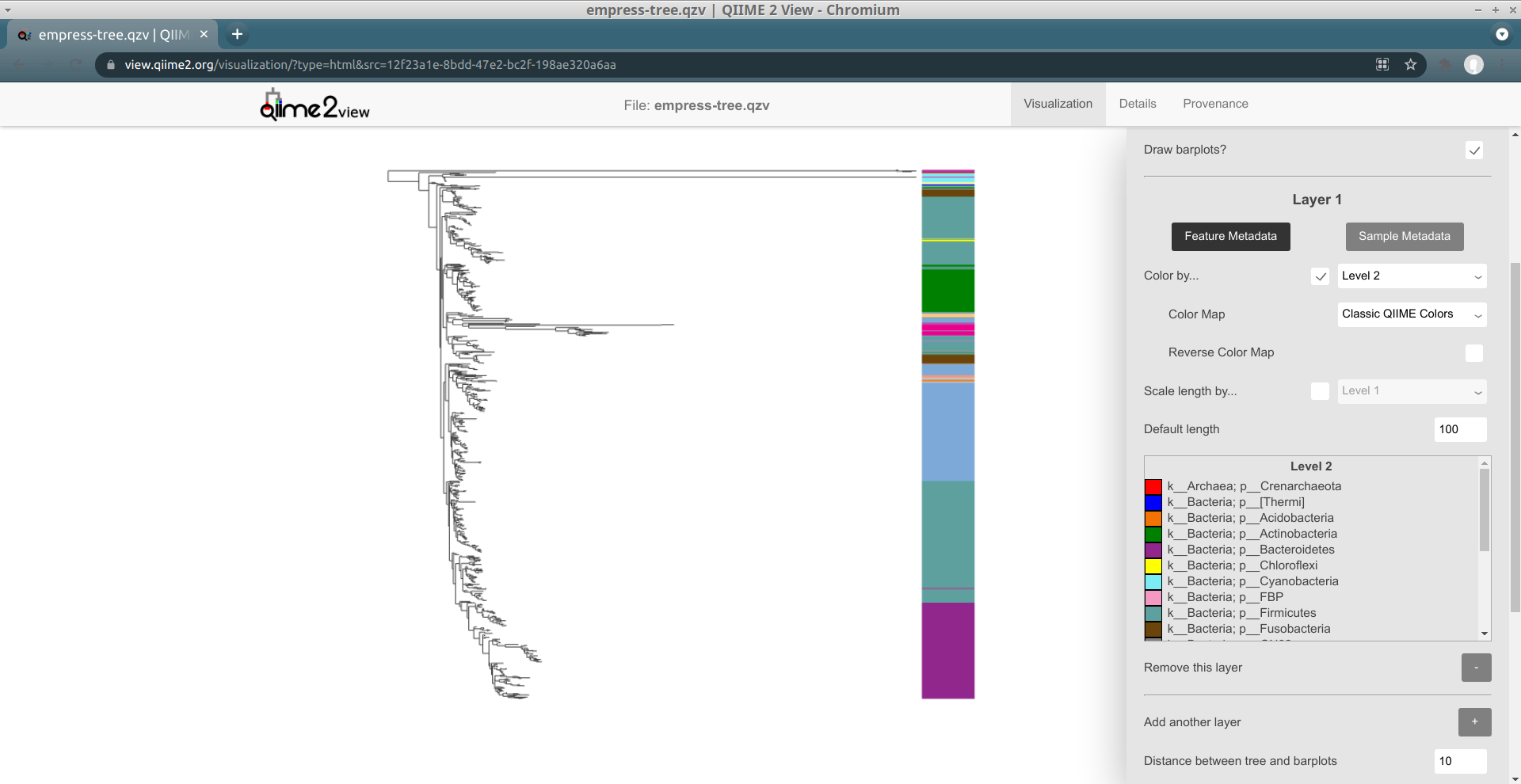
These patterns should look familiar -- this is the same information as we showed when coloring the tree by feature metadata earlier. We can confirm this by trying out feature metadata coloring by Level 2 again, using the same Classic QIIME Colors color map (see the "Exploring groups of features" section above for a refresher on how to do this):
Since both the node colorings and the barplot layer are now showing the same information (Level 2), this display is a bit redundant (although it is reassuring :). Let's try taking things down a level, and adjust our barplot layer to show the Level 3 feature metadata field (a.k.a. the class-level taxonomic assignments). To do this, adjust the drop-down menu next to the Color by... box (under the Layer 1 header in the Barplots section, not in the Feature Metadata Coloring section) to go from Level 2 to Level 3, and then click the Update button again.
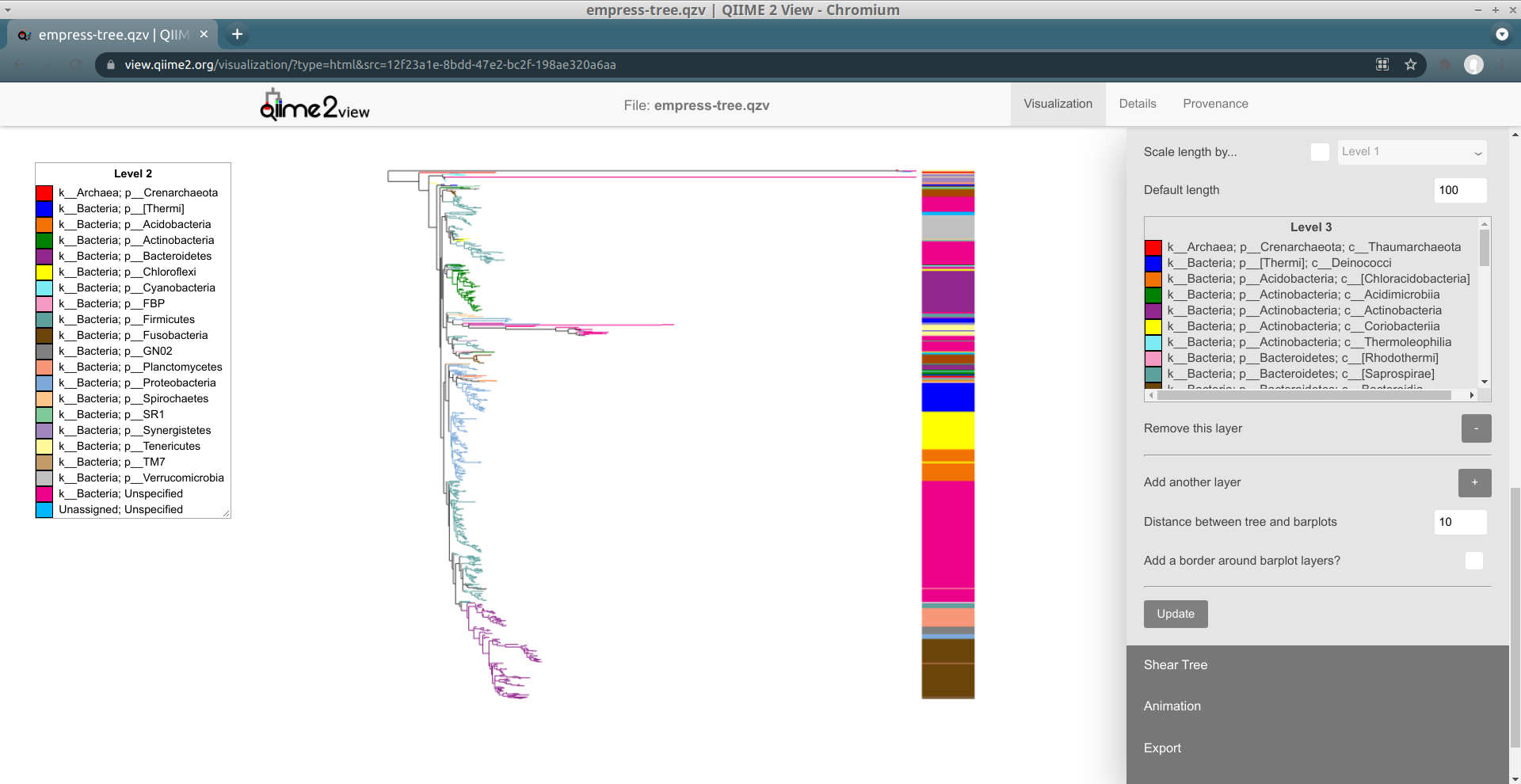
Things still seem mostly the same as before, but some of the large groups of phyla have now been split up into collections of different classes. Notice how the magenta-colored class is present at multiple "clusters" throughout the tree: are all of these the same class? We can tell from the legend for this layer (under the heading Level 3) that there is only one class colored magenta here, k__Bacteria; p__Firmicutes; c__Clostridia.
So, these magenta classes are all Clostridia. Does it make sense that representatives of this class are spread out throughout the tree so much? Unfortunately, yes, since Clostridia are -- to quote Wikipedia -- "a highly polyphyletic class." (As an exercise, we recommend trying out adding on extra barplot layers for lower levels of taxonomy -- order, family, genus, etc. -- and seeing how things change.)
Barplots of sample presence information
Up until now, we've just been working with a single "barplot layer." We can add on more layers if we want -- this will let us visualize additional tip information alongside the layer we have that currently shows Level 3 information. To add a new layer, click on the + button (with the label Add another layer). Now, click Update again to see what this new layer looks like.
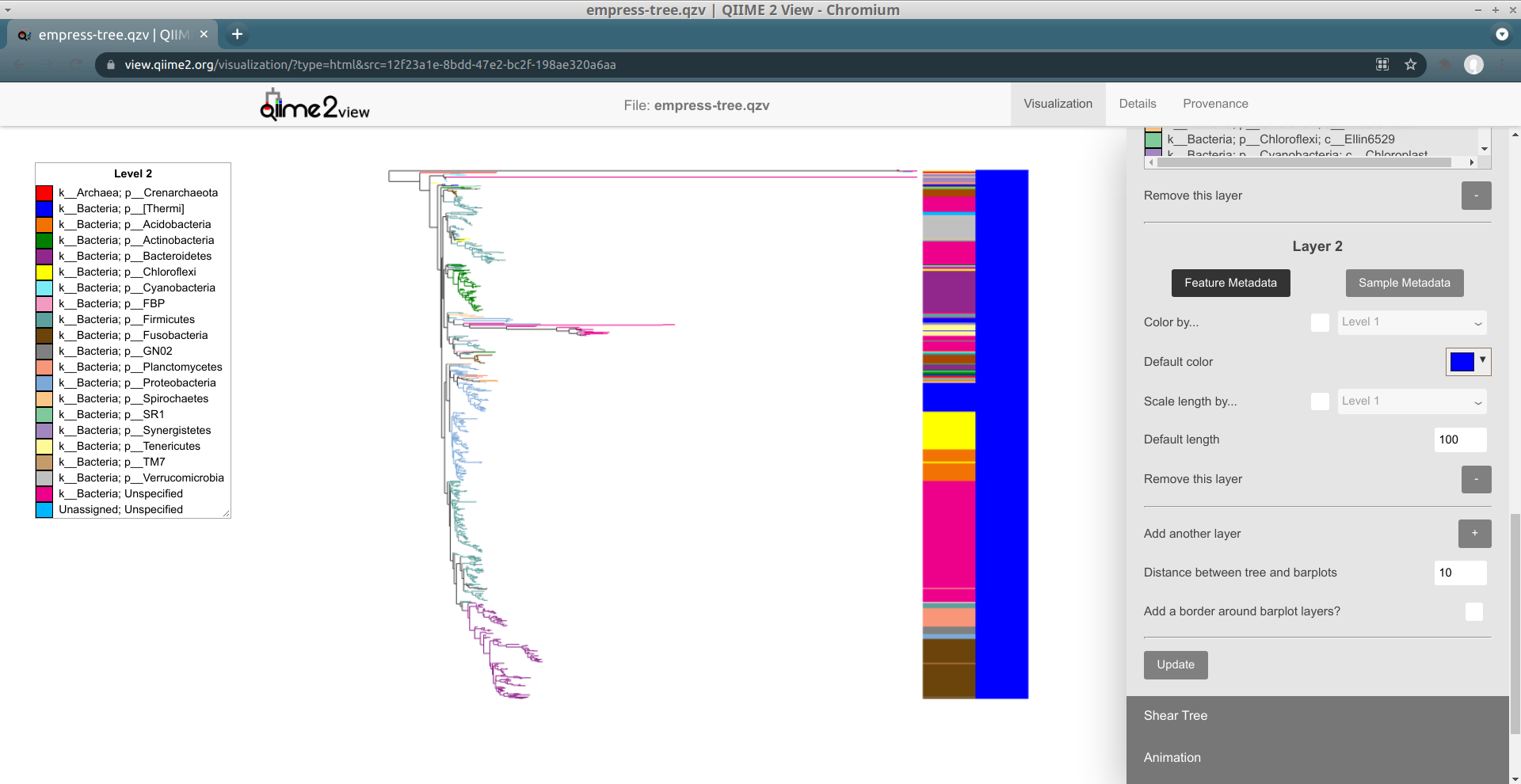
We have a new layer to work with!
One thing we might be interested in doing is seeing what types of samples contain each tip. This is possible using the Sample Metadata Coloring functionality described above, but this only lets us see information about tips that are unique to a given sample metadata category -- and in practice many tips are often shared between multiple metadata categories, complicating things.
Let's revisit our analysis above of which tips are unique to which body sites in this dataset -- now, we'll instead be asking the related question of "which body site(s) are the tips in this dataset most frequently seen in?" To investigate this, we'll use our new barplot layer to show this information.
In order to do this, we'll need to change our new layer (Layer 2) from a feature metadata layer to a sample metadata layer. You can do this by clicking on the Sample Metadata button underneath the text Layer 2. The controls available for this barplot layer should change; in order to show sample presence information for body sites, change the Show sample info for... drop-down menu to body-site. Try clicking Update to see what our new Layer 2 looks like.
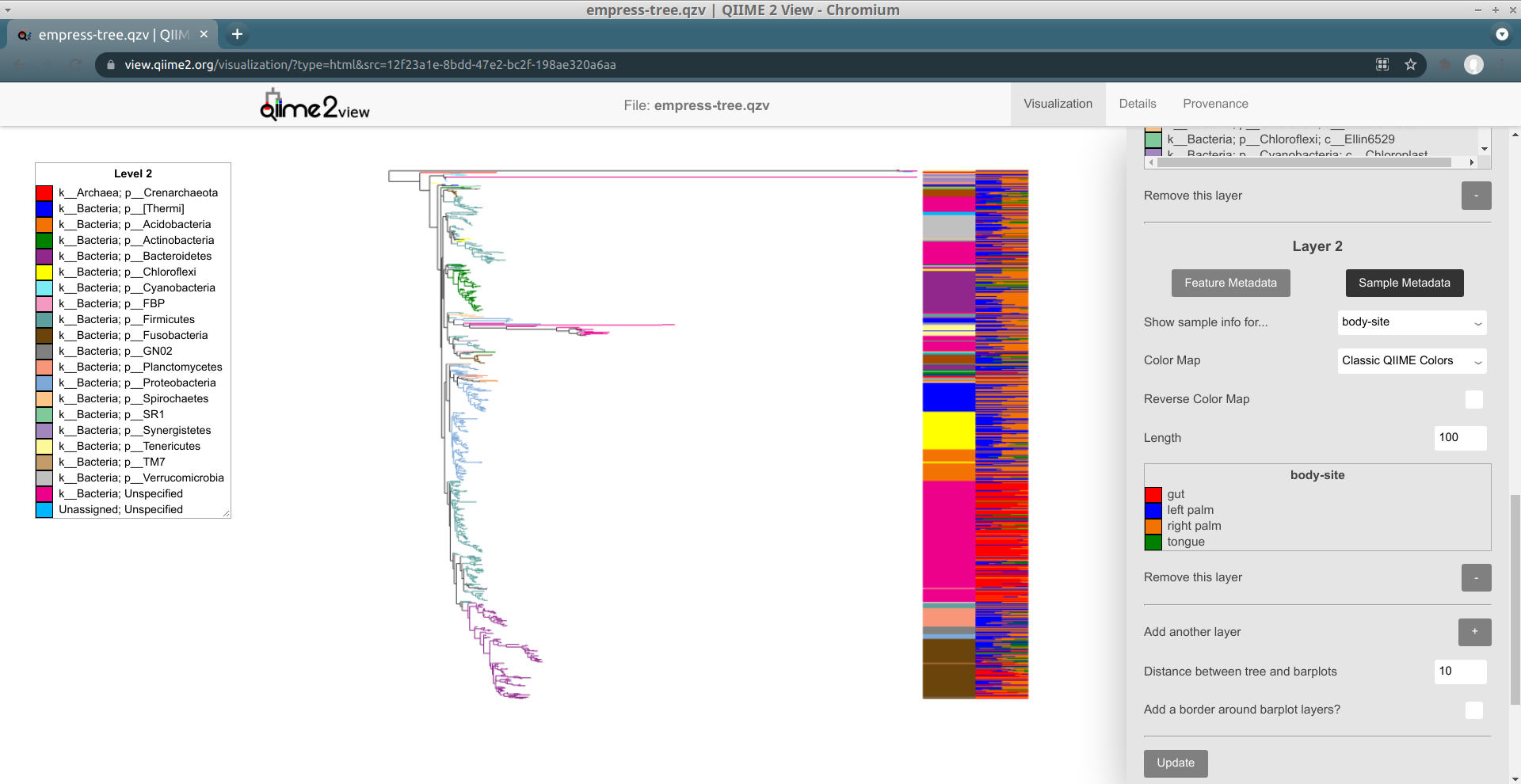
Layer 2 now shows a stacked barplot for each tip, based on the proportions of sample groups containing a given tip. As with layer 1, the colors are described in this layer's legend. When we zoom in, we can see things in detail:
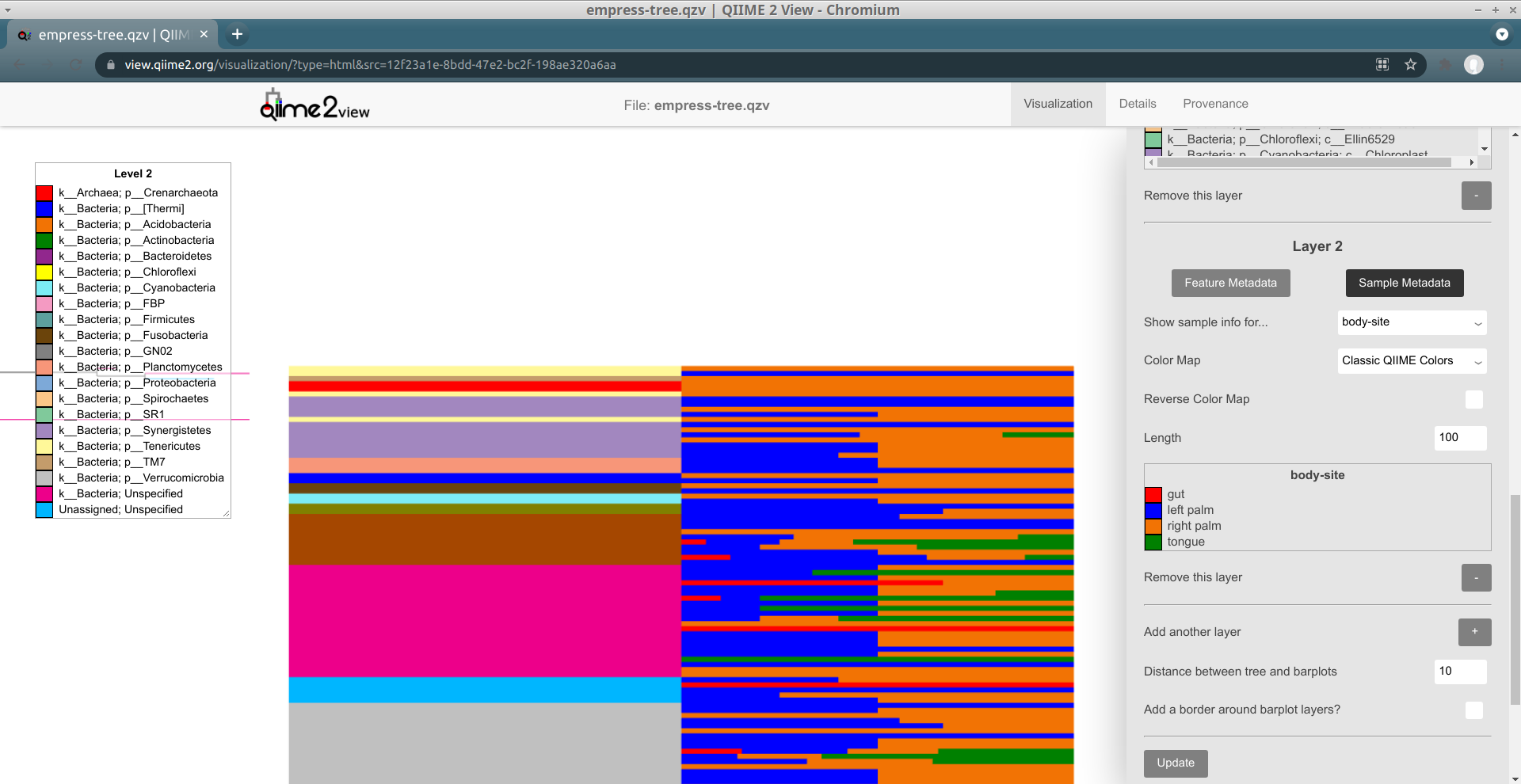
The top-most tip is only present in right palm samples (colored orange), the second-from-the-top tip is only present in left palm samples (colored blue), and so on. The length taken up by a "block" for a given tip is proportional to how many samples of that type contain the tip (relative to the total number of samples containing the tip; it's not absolute).
These sample metadata barplots should match up with the Sample Presence Information -- try clicking on the top-most tip, 35bfc371d940cffdc527b7b4dc954456. We know from the barplot that this tip is only present in one right palm sample, and the Sample Presence Information summary by body-site for this tip confirms this.
Barplots of continuous feature metadata
Although drawing barplots of "categorical" feature metadata (like taxonomy annotations) can be useful, often we'd like to display barplots of continuous feature metadata. This can be useful for many types of information -- for example, importance scores, Songbird/ALDEx2/etc.-style feature differentials, and taxonomy annotation confidences.
Here we'll add on another layer describing the Confidences of the taxonomy
annotations in this dataset. (See this thread for details about interpreting these values.)
All of the confidence values in this dataset are numeric, but we don't have to interpret them as numbers. Let's see what it looks like if we try to use a "categorical" (a.k.a. "discrete") color map for this field. Click on the + button to add a new layer, check the Color by... box, and select Confidence. Click the Update button.
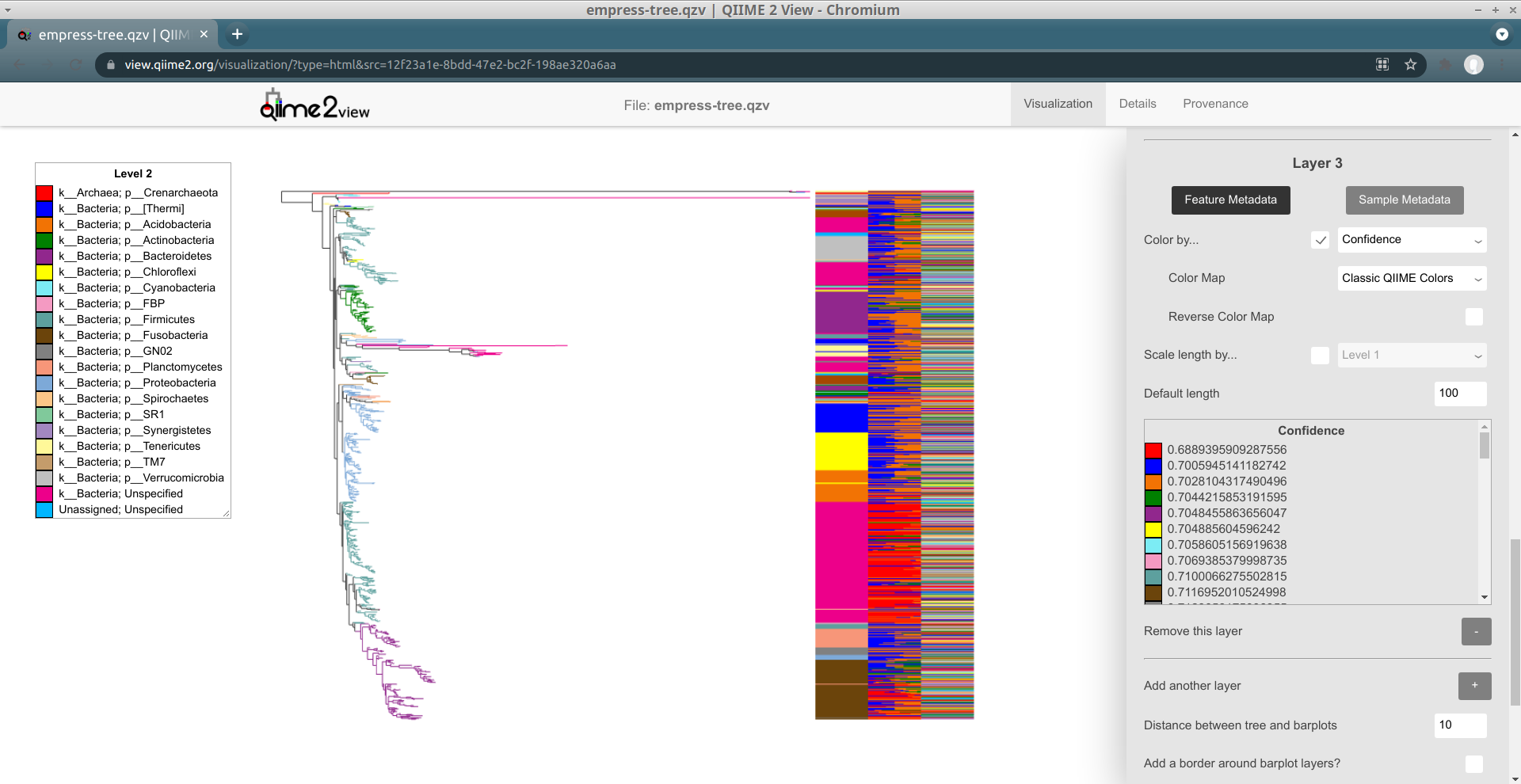
Try scrolling through this layer's legend. It should be clear that this color map (Classic QIIME Colors) does not make sense for this field -- although the confidence values are correctly sorted in ascending order, the actual color assignments are meaningless.
Let's try changing away from this discrete color map to a "sequential" one.
Select Viridis from the Color Map drop-down menu, and click Update.
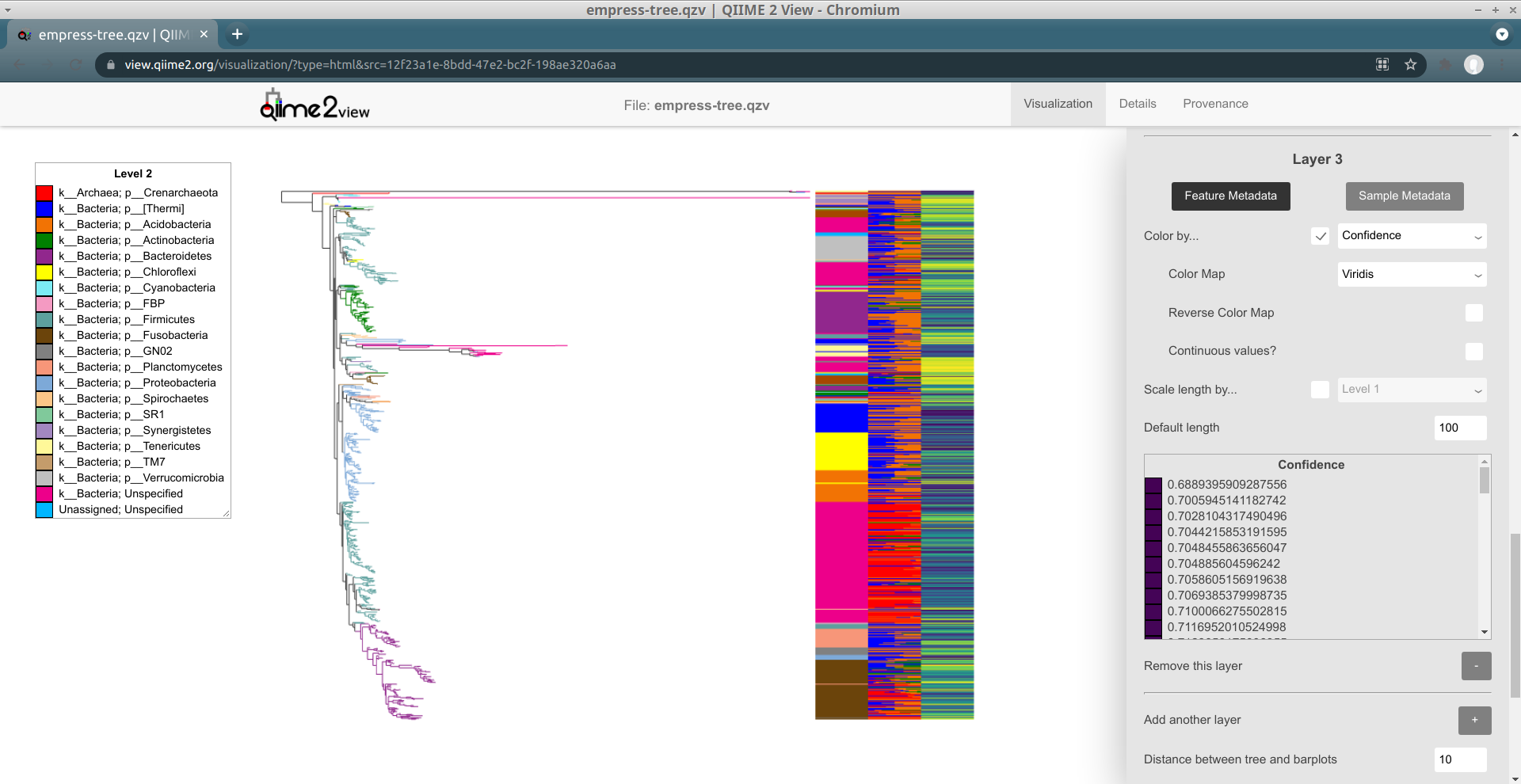
Try scrolling through the legend now. Things should be a bit clearer -- there's
a gradient from purple to yellow that seems to align with the Confidence
values. However, this color map is still not considering the actual numeric
values of these Confidences: only the relative positions of these values are
taken into account. Notice how there are so many Confidence values with values
greater than 0.99, and that in spite of this the colors assigned to these
values vary pretty wildly (even though the minimum Confidence is around
0.68)!
We can fix this by checking the Continuous values? box (it shows up when a
sequential or diverging color map is selected). Try doing that, and then click
Update one last time for now:
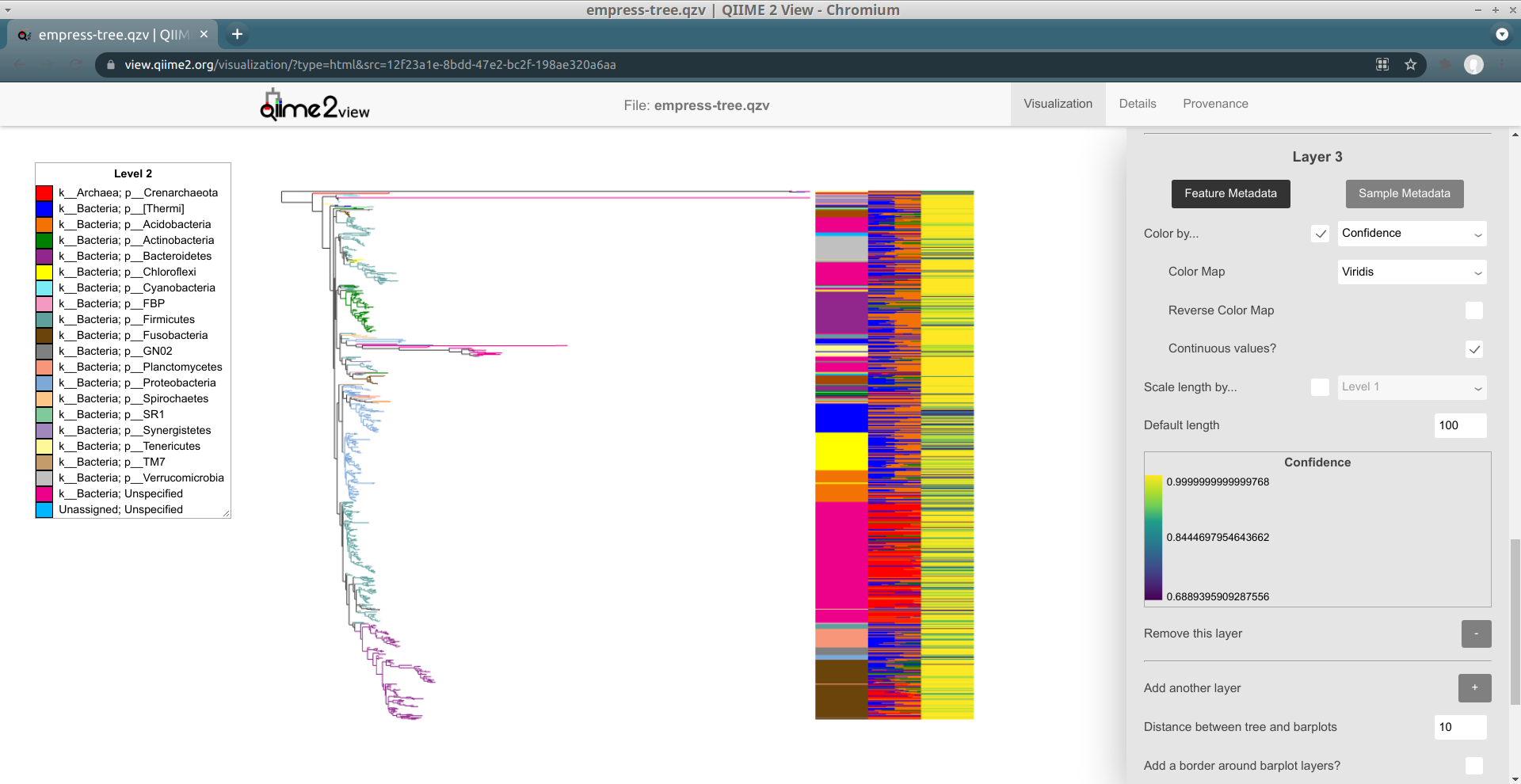
Now, colors are assigned based on Confidences as we might expect, using
linear interpolation.
This was a brief introduction to some of the barplot functionality available in Empress. There's a lot more that hasn't been documented here -- scaling bars' lengths by a continuous feature metadata field, adjusting the default colors or lengths of bars, and so on. We encourage you to try things out; feel free to contact us if you have any questions!
Exporting Plots
Once you are done customizing your tree, you can export the current visualization of the tree as an SVG or PNG file by going to the Export section in the main menu and clicking on Export tree as SVG or Export tree as PNG. You can also export the legend(s) used for tree and/or barplot coloring, if applicable, using the Export legends as SVG button.
Note that SVG export will always include the entire tree display, while the contents of the PNG export will change as you zoom / pan the tree.
Empire plots! Side-by-side integration of tree and PCoA plots
Now that you are familiar with basics, let’s try something a bit more advanced. One of the unique features of Empress is its ability to integrate a tree plot with an Emperor ordination plot and visualize them side-by-side (we've taken to calling these Empire plots).
To achieve this, we can provide a QIIME 2 artifact of the type PCoAResults to qiime empress community-plot. (Note that although the type is PCoAResults this can be any ordination matrix; it's totally possible to visualize the results of PCA, or even a biplot, here instead.)
Biplots include feature loadings represented by arrows that describe explanatory variables (in this case, ASVs) in the dataset. Here, we'll visualize a PCoA biplot made using the qiime diversity pcoa-biplot plugin. This biplot was computed using Unweighted UniFrac distances. (This functionality is also compatible with DEICODE biplots, of course.)
To visualize an Empire plot (both the tree and this PCoA biplot together), run the following command:
qiime empress community-plot \
--i-tree rooted-tree.qza \
--i-pcoa biplot.qza \
--i-feature-table table.qza \
--m-sample-metadata-file sample_metadata.tsv \
--m-feature-metadata-file taxonomy.qza \
--p-filter-extra-samples \
--p-number-of-features 10 \
--o-visualization empire-biplot.qzv
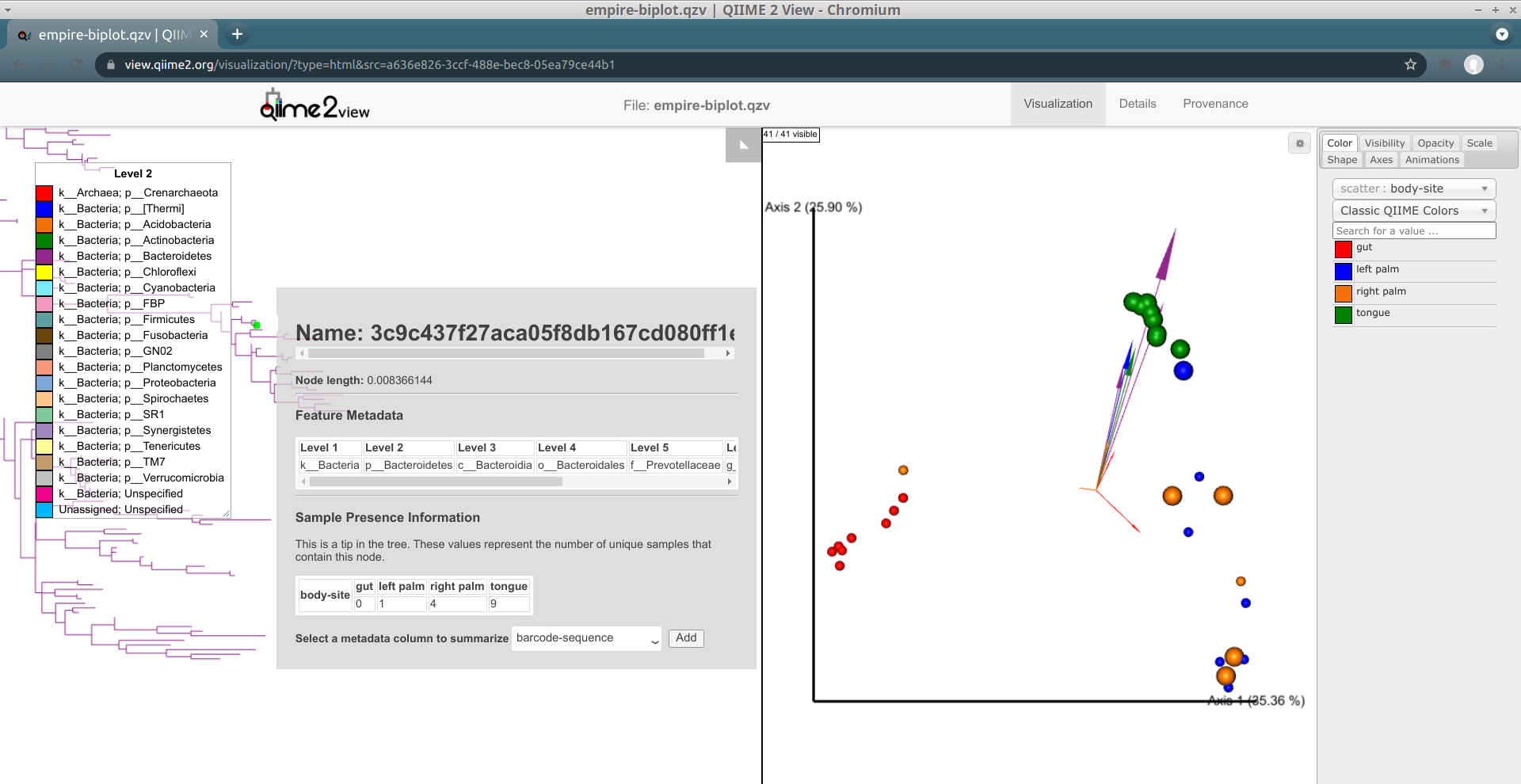
Load the new Empire plot. Here we see the Empress plot as before on the left, and on the right is an Emperor PCoA biplot. If you are unfamiliar with Emperor plots, you can learn more about them here. Briefly, each individual circle represents a single sample’s microbial community and the distances between these circles corresponds to the Unweighted UniFrac distance between them in a reduced dimensional space. The top 10 explanatory features are shown as arrows alongside their feature IDs. The number of features that is shown on the biplot is determined by the --p-number-of-features parameter.
At first, the plot may look a bit messy. For clarity, let’s remove the long feature ID labels. Right click anywhere on the Emperor plot and select Toggle label visibility. Next, in Emperor, from the main menu click on Select a color category and select body-site under the scatter subheading. Now our samples are color-coded based on their body site origin. Notice the clear clustering of these sample-types. Next, click on the same drop-down menu and this time under the biplot subheading select Level 2. Now we can see the top explanatory features (arrows) colored by their phylum-level classification. Switch over to Empress, change the plot layout to Circular, and set the Feature Metadata Coloring to Level 2 also. Minimize the menu bar to fully appreciate the plots!
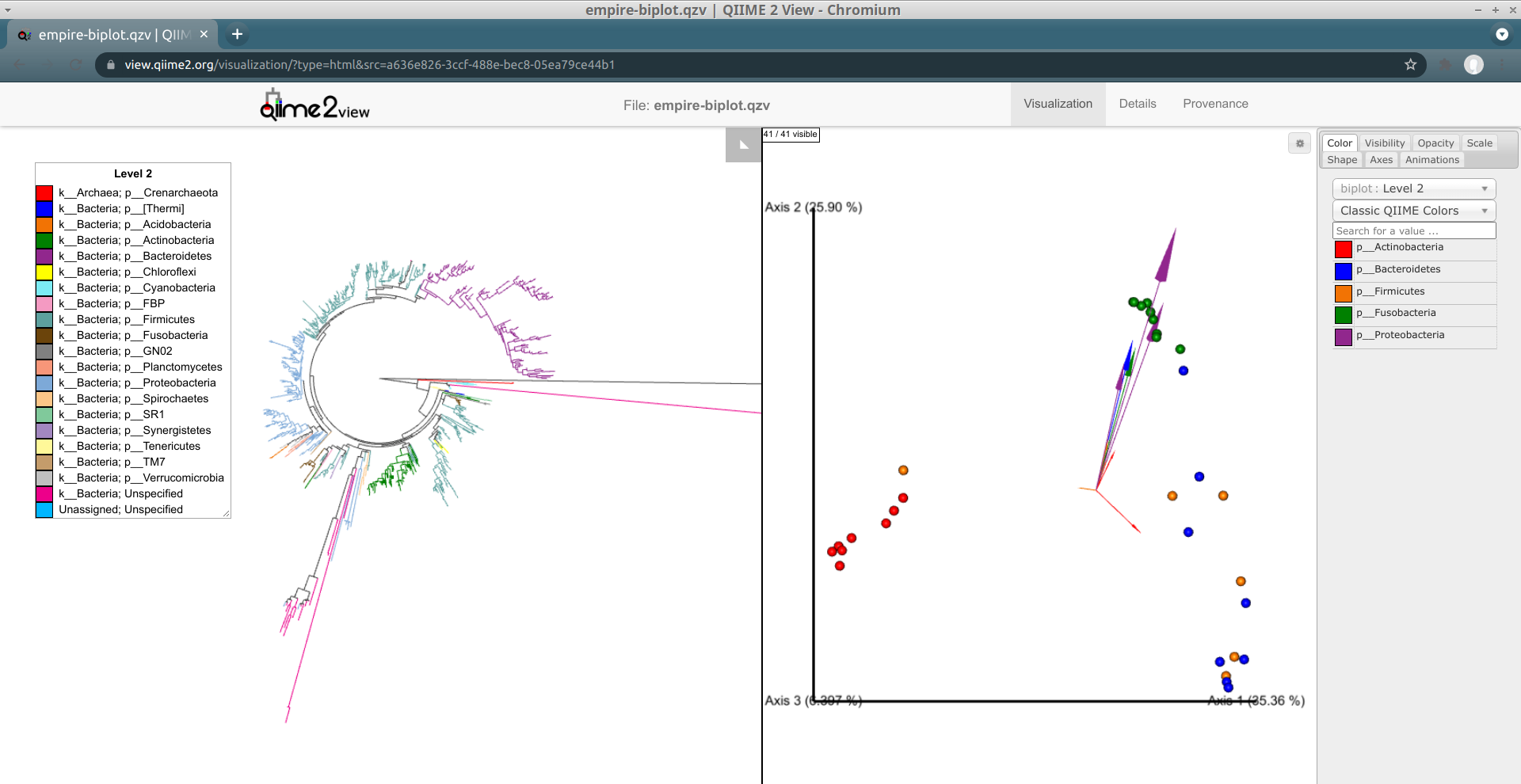
(Note that the tree and arrow colorings don't necessarily match up between Empress and Emperor—for example, in the screenshot above Actinobacteria-phylum arrows are colored red in Emperor but Actinobacteria-phylum nodes are colored dark green in Empress. If you'd like, you can change the arrow colors in Emperor to match the colors Empress assigned. Making this easier is on our radar.)
Interacting with Empire plots
Looking at our Emperor ordination plot (on the right), we see a single feature classified in the phylum Actinobacteria (a small red arrow) that is associated with the palm samples. It's pointing towards the bottom-right of the ordination, when looking at it in the default camera position.
Click on this arrow (you may have to zoom in a bit in Emperor to do so). Two changes will automatically occur:
- In Empress, the plot will zoom in on the node corresponding to this feature and open a menu. From here, you can explore the details for this feature (which it turns out was classified as Corynebacterium sp.) further, just like we did before.
- On the Emperor plot, the samples that contain this feature will be enlarged, clearly highlighting them in contrast to the other samples that do not contain this feature.
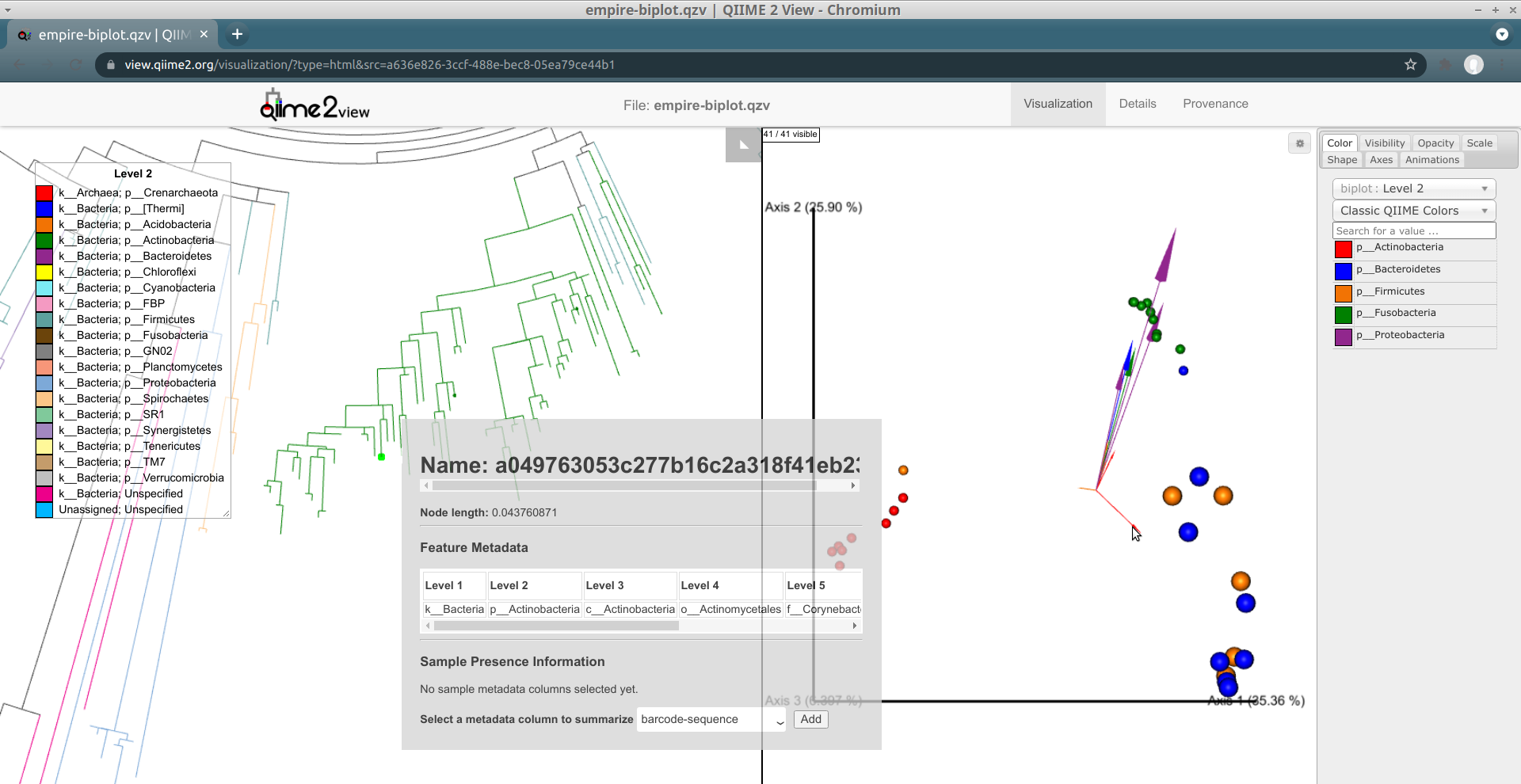
This interaction between Empress and Emperor can go the other direction. Selecting a node on the Empress plot will enlarge the samples in Emperor in which that feature is present.
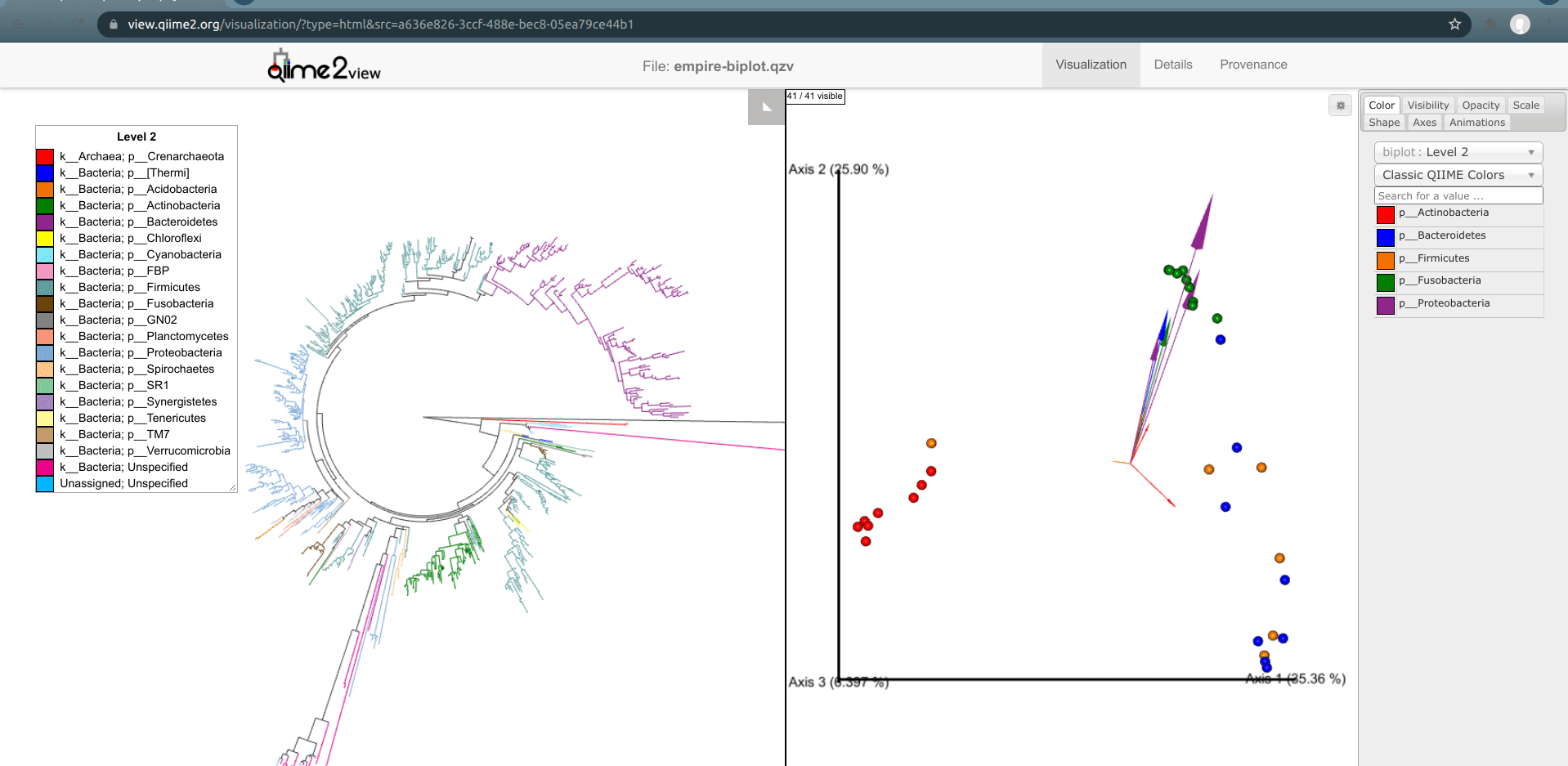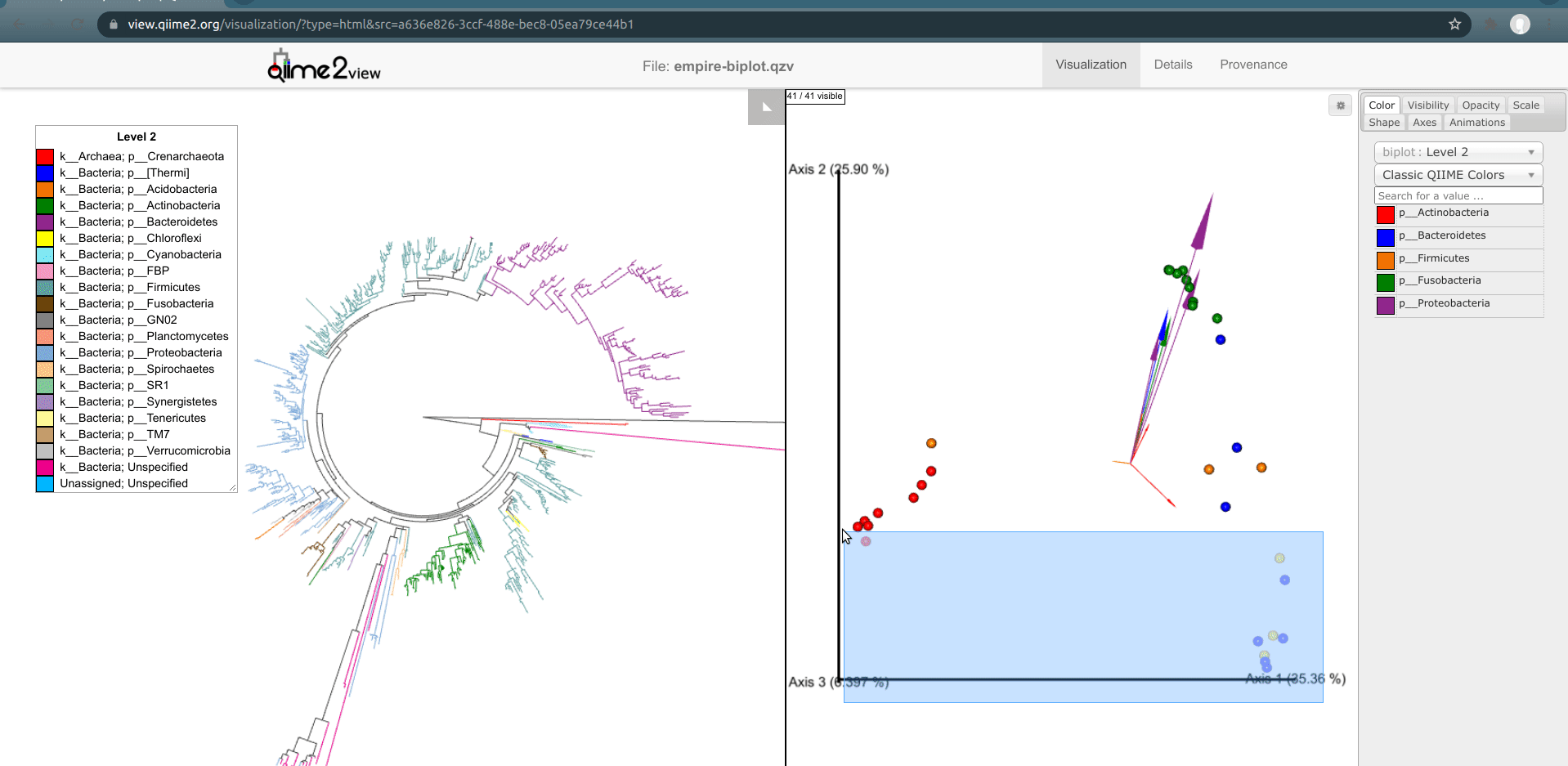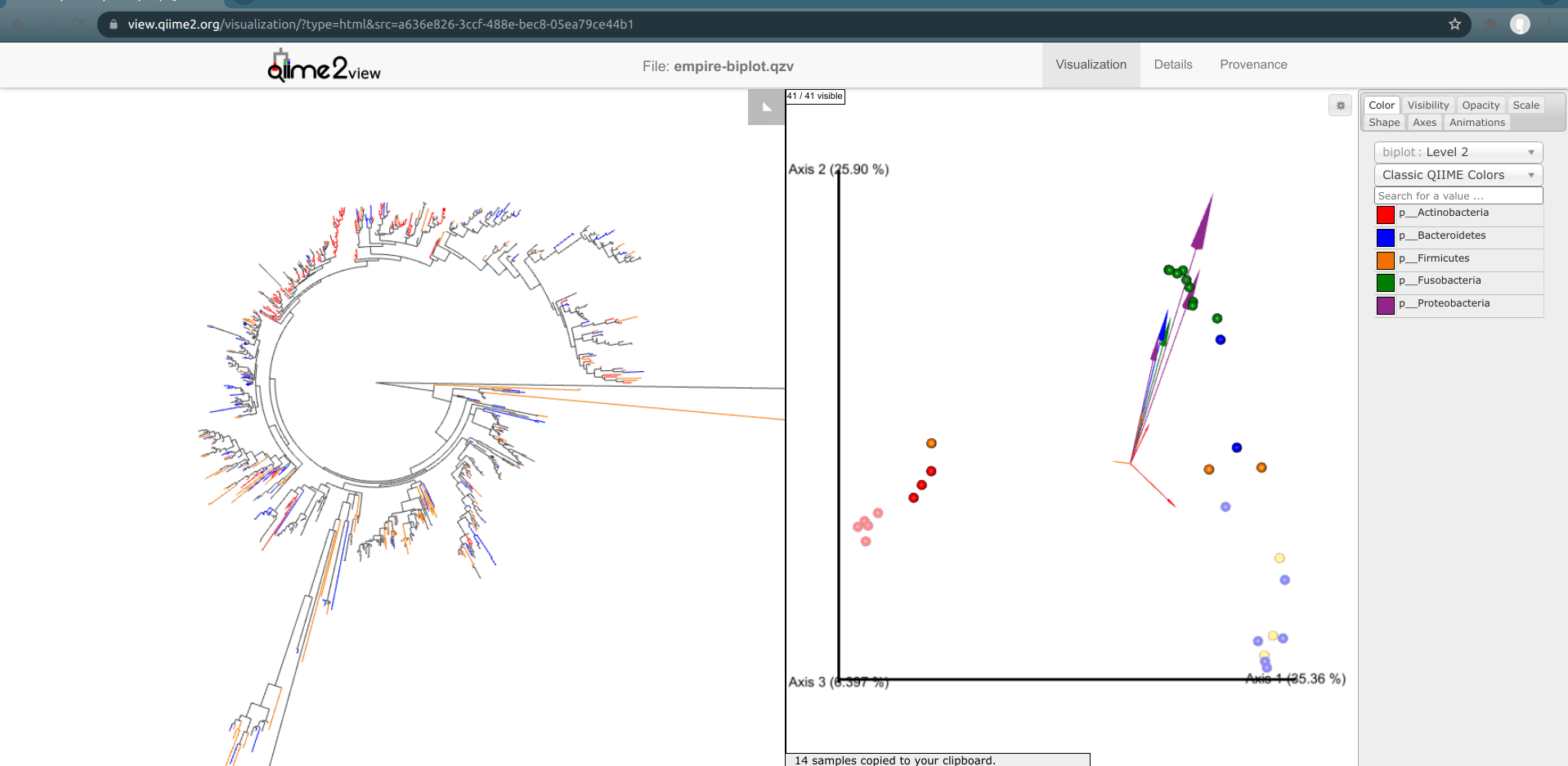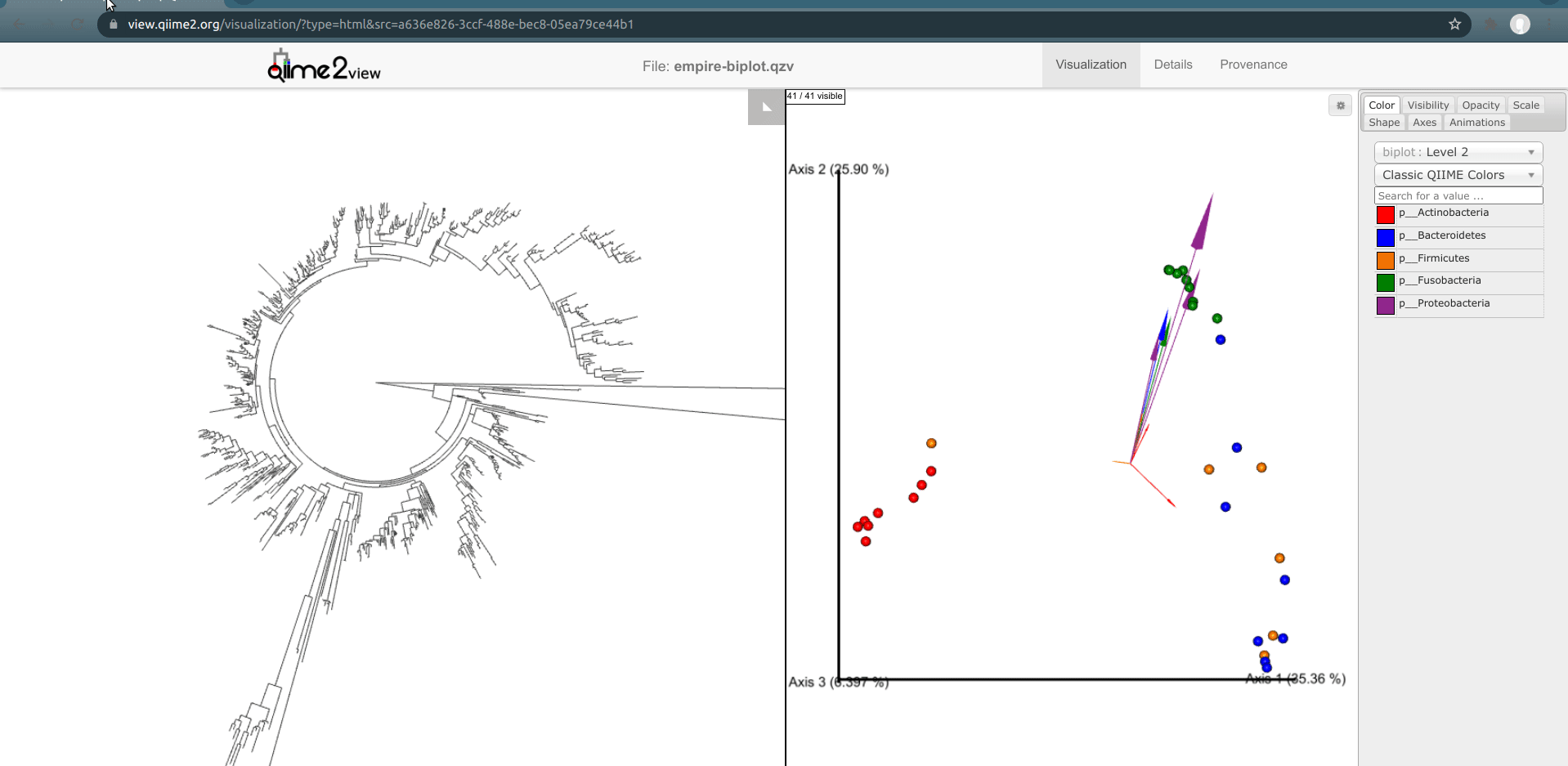
Another way to explore our data is to select samples on Emperor and look for the corresponding features present in these samples in Empress. In Emperor, hold the shift button and draw a box around a sample. The Empress plot will now temporarily highlight the branches corresponding to that sample. If you select multiple samples from different body-sites, Empress will only highlight the branches/nodes that are unique to those sample types. The shared branches remain uncolored. Let’s see how we can utilize this function in our dataset.
You may have noticed that in the Emperor plot one of the Right Palm samples is strangely clustering closer to the gut samples rather than the other palm samples. On Emperor, select some of the gut samples as well as some of the palm samples from the right hand side, taking care to not include the outlier palm sample on the left. On the Empress plot you will see several branches light up as either red, orange, or blue. These colors represent the unique features found in only that body-site; shared features are left uncolored.
Once the samples have been deselected (within a couple of seconds), select the outlier palm sample + one of the gut samples. What do you notice? You’ll see that comparatively few unique red or orange branches light up, suggesting that this sample shares many more features with the gut samples than the other palm samples.*
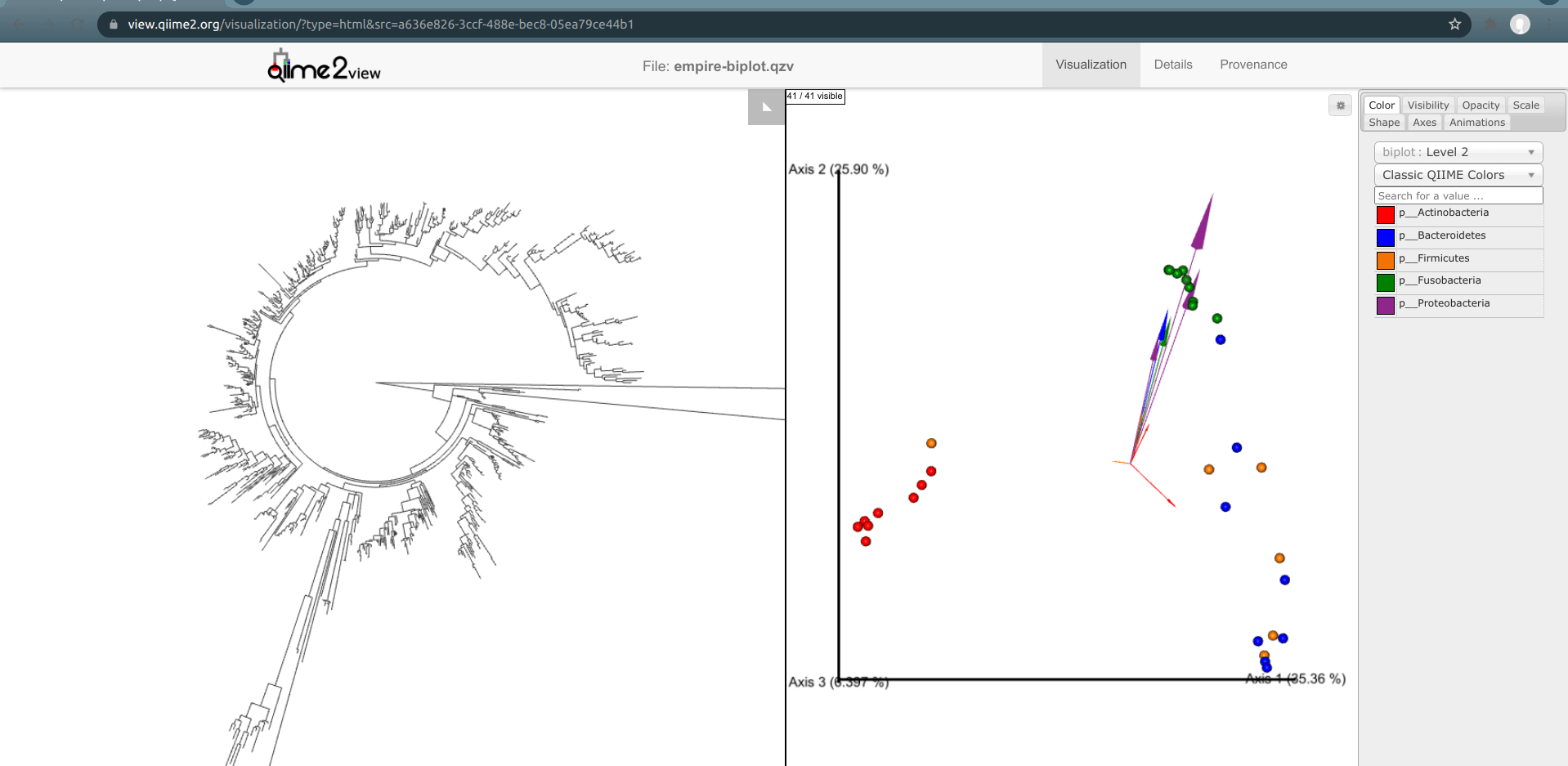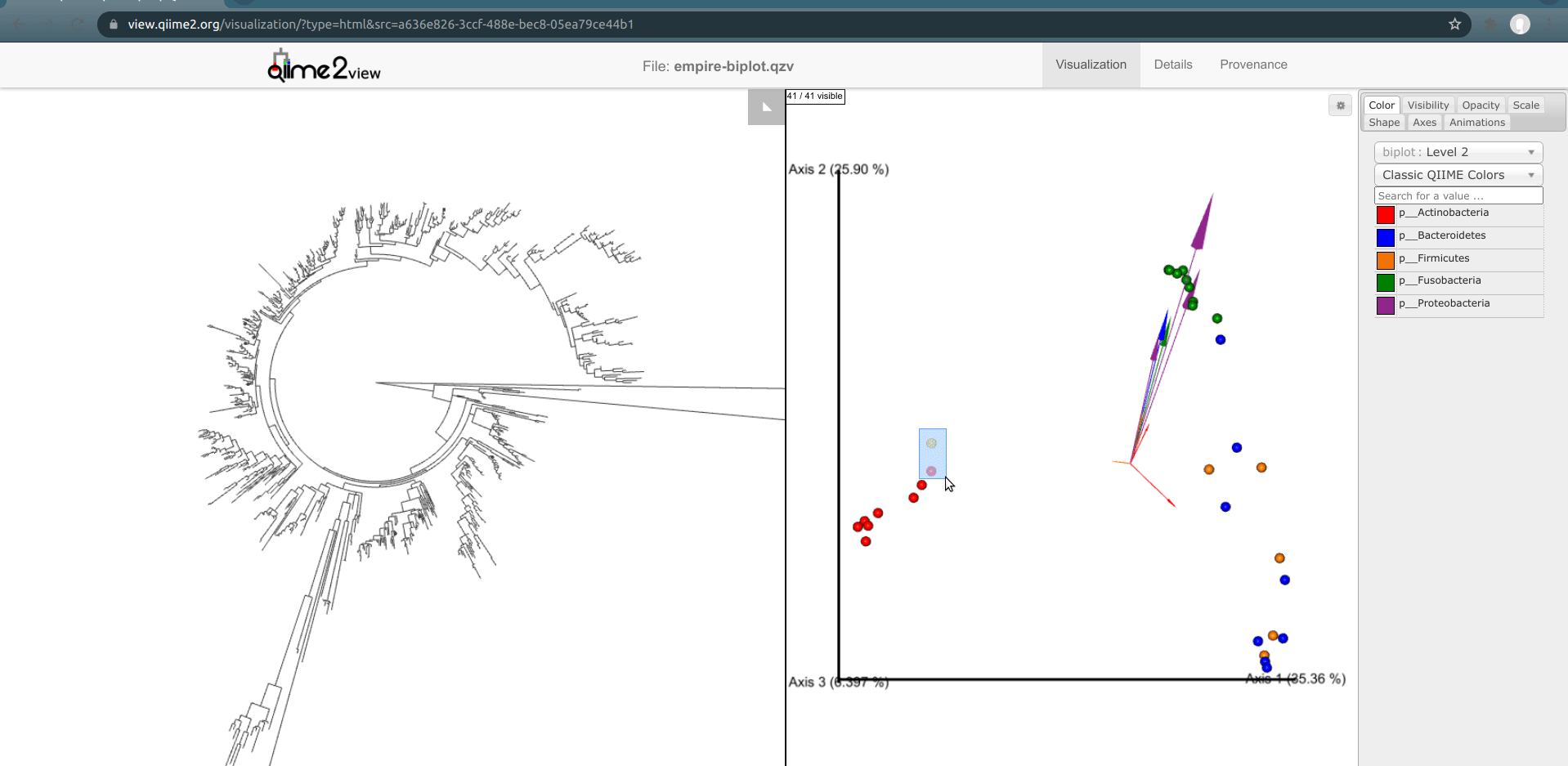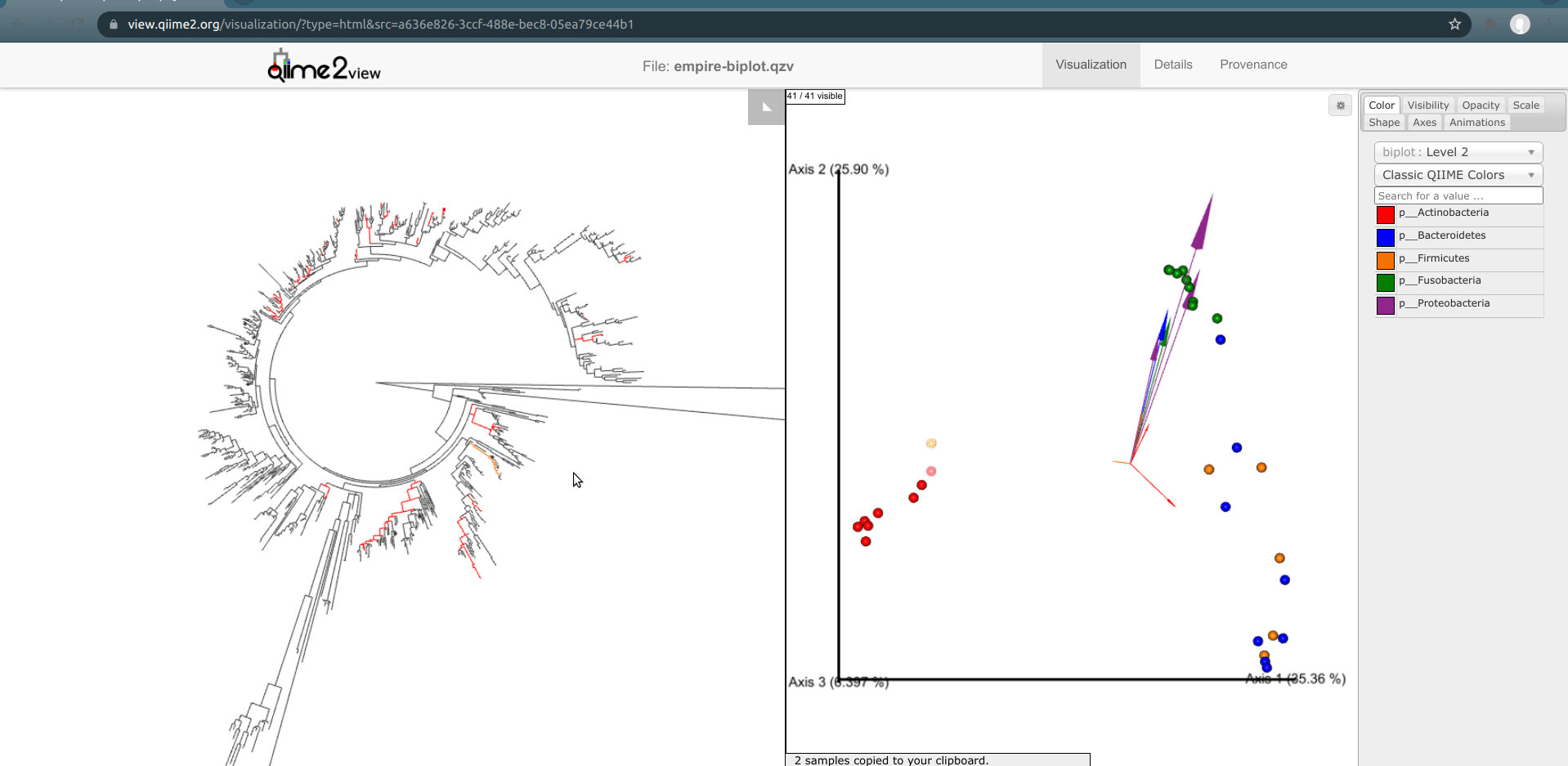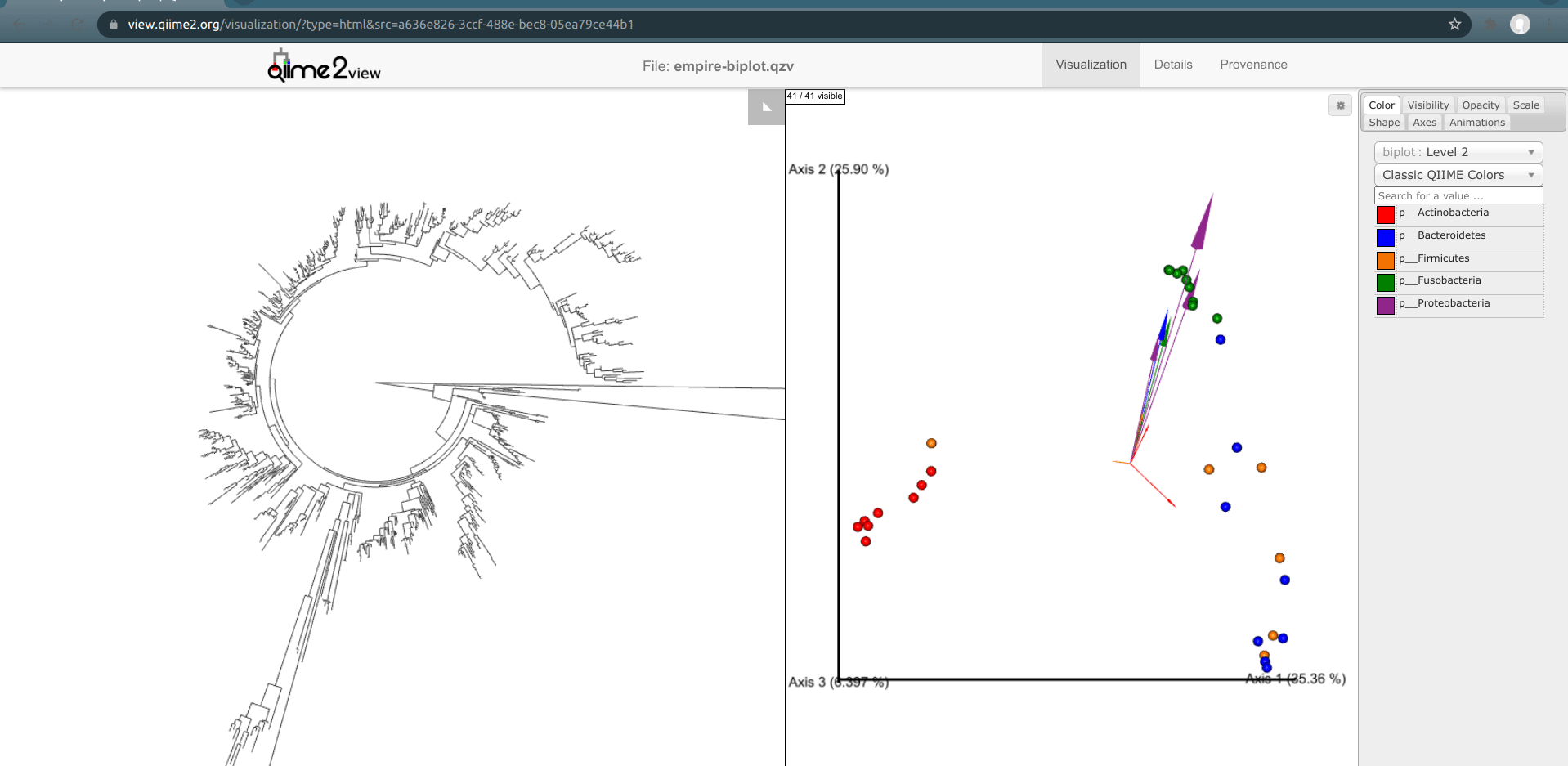
This is a good example of when your data can tell you something about your metadata that you may have missed. In reality, in this experiment, this palm sample was in fact mislabelled by accident.
* Note that this intuition relies partly on the Ignore absent tips? setting in Empress, which impacts how tips present in none of the selected samples impact the tree coloring. By default, the parent nodes of these tips are allowed to be colored if another one of their descendant tips actually is present within the selected samples; but if Ignore absent tips? is disabled, then these "absent" tips will force their parents to remain uncolored.
The integration between Empress and Emperor can go even further than this. Rather than selecting a group of samples manually, we may want to just select all samples in a certain group (e.g. for this dataset, all gut samples at once), to show which features are present in these samples. This can be done by double-clicking on a sample coloring category in Emperor, as shown below:
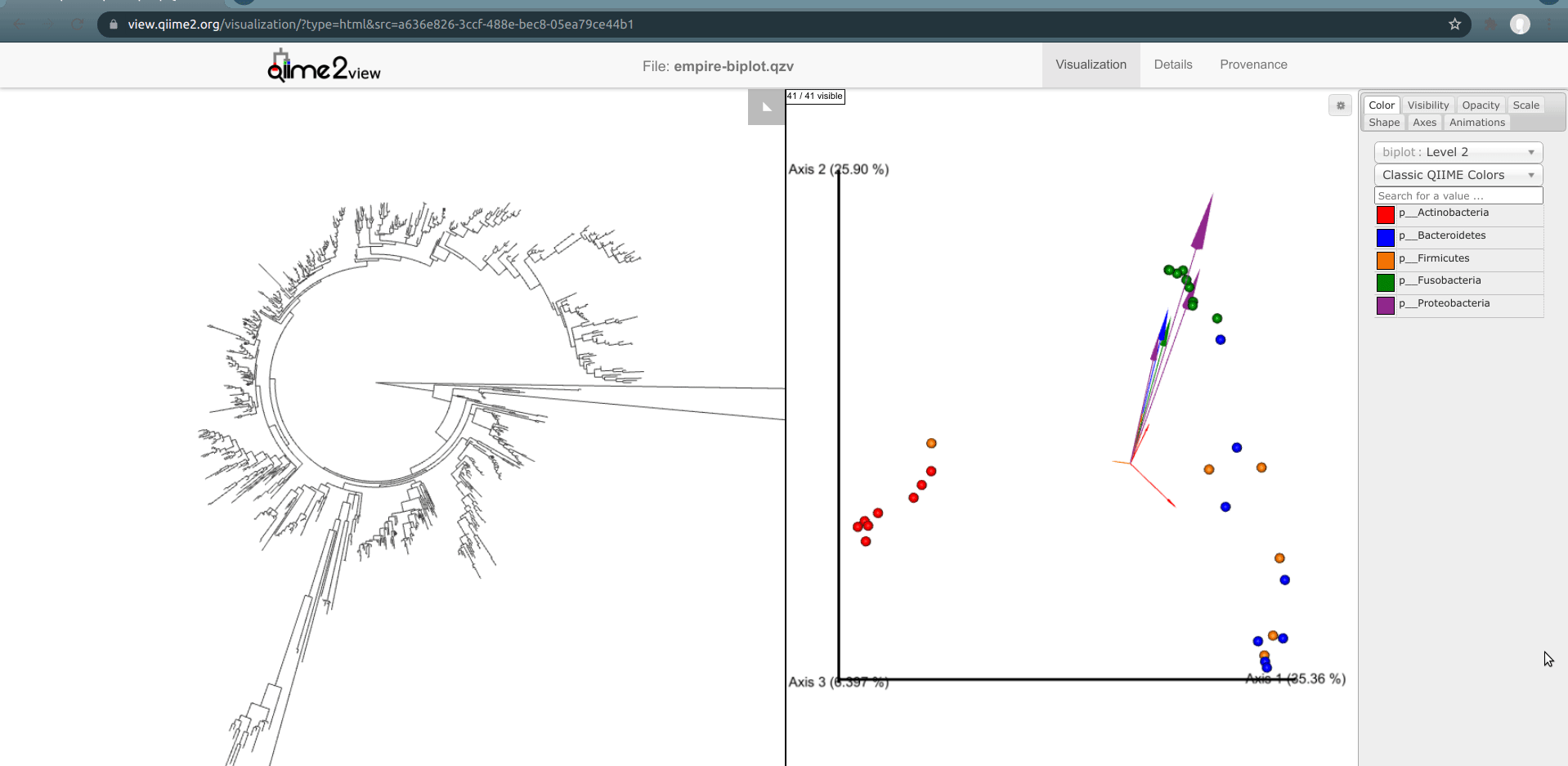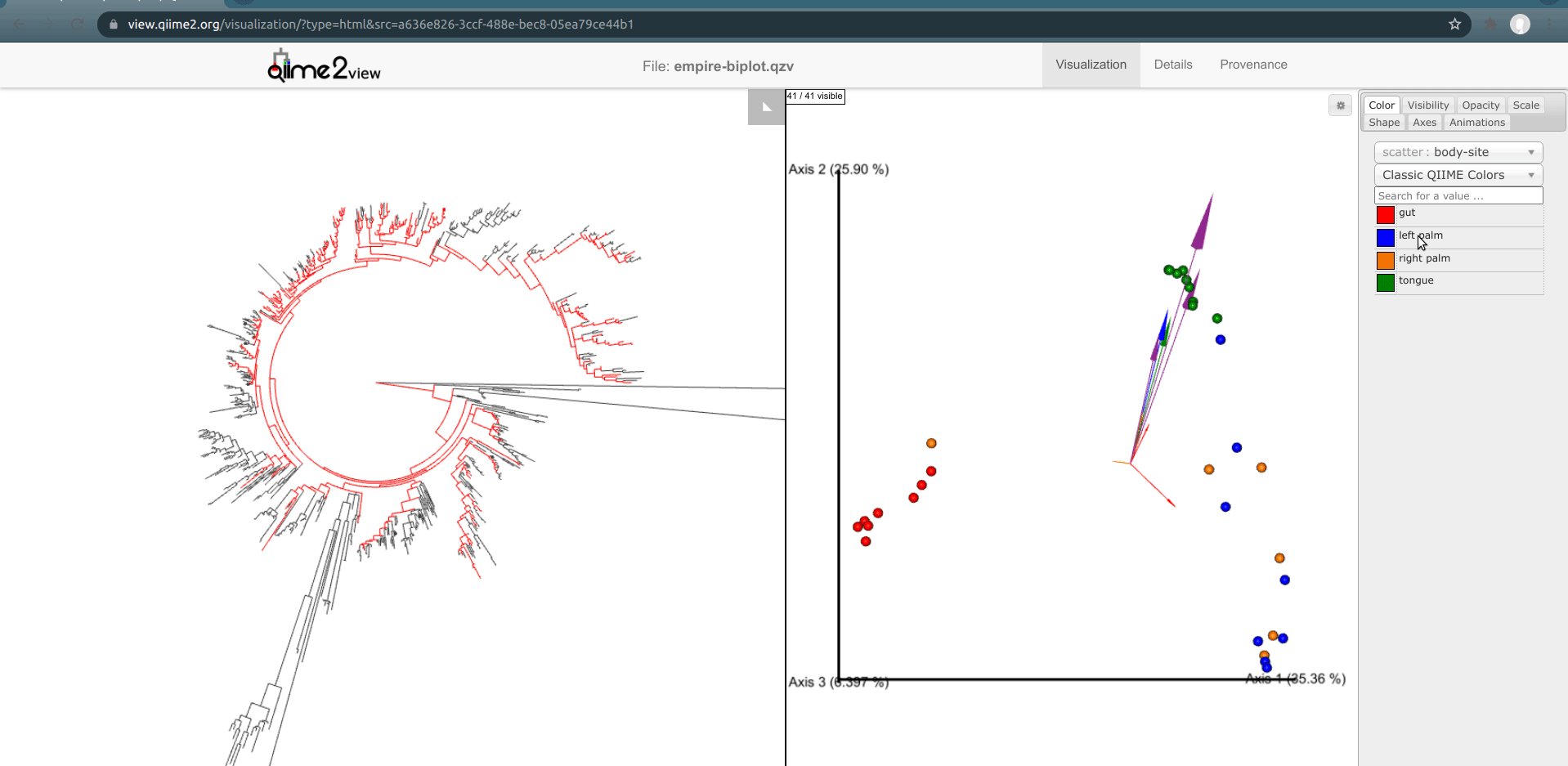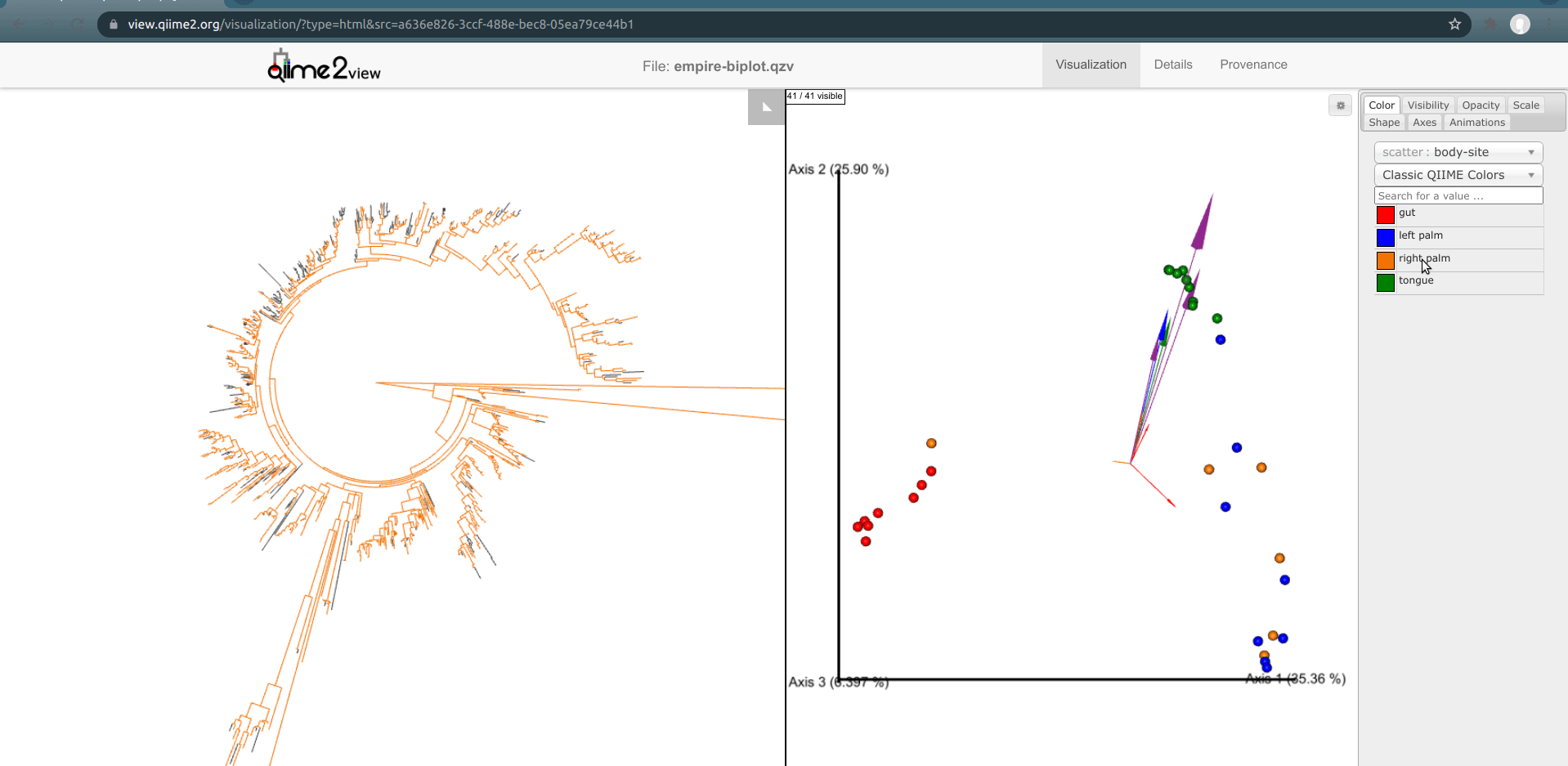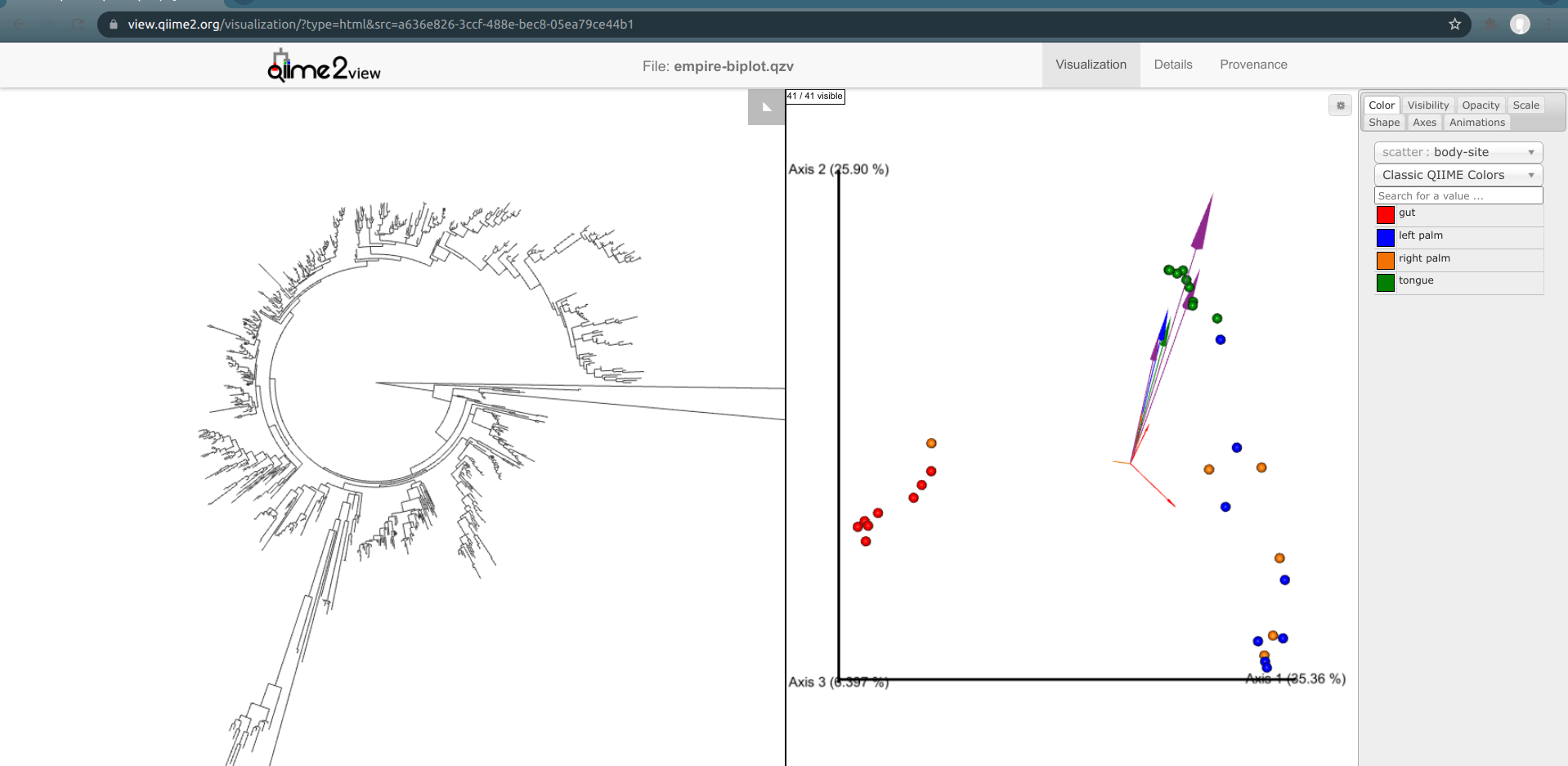
This makes it easy to get a quick glance at which parts of the tree are "used" within a certain group of samples. (If you have a hard time viewing certain colors on the tree—for example, distinguishing the blue color for left palm samples from the default dark-gray node color—you may want to adjust the sample group colors in Emperor.)
Additional Considerations
Providing multiple metadata files
QIIME 2 allows you to specify multiple metadata files at once by just
repeating --m-feature-metadata-file (or --m-sample-metadata-file). For
example, we may want to visualize feature importances on a tree
in addition to taxonomic annotations:
qiime empress community-plot \
--i-tree rooted-tree.qza \
--i-feature-table table.qza \
--m-sample-metadata-file sample_metadata.tsv \
--m-feature-metadata-file taxonomy.qza \
--m-feature-metadata-file feature_importance.qza \
--o-visualization empress-tree.qzv
However, what QIIME 2 will do internally (as of writing)
is filter the metadata to
just the entries contained in all of the input metadata files. So, in the
example above, if the feature_importance.qza file only has entries for a
couple of features (compared to the taxonomy.qza file), then the feature
metadata Empress receives will be limited to just the features contained in
both the feature importance and taxonomy metadata files -- which will mean that
less taxonomy information will be available in the Empress interface!
In the interim, the way to get around this (and to include multiple sources of
feature or sample metadata in Empress) is to merge metadata yourself before
creating an Empress visualization. Of course, you'll need to determine what
value(s) to assign to indicate that a given entry is "missing"; for
quantitative metadata, NaN or an empty value are both reasonable options.
Merging metadata files should be doable in many different programming languages or spreadsheet tools; see this GitHub issue for some example Python code that does this.
Filtered vs. raw table?
When your ordination was created from a subset of your original dataset (e.g. the feature table was rarefied, or certain low-frequency features or samples were otherwise filtered out), we recommend that you carefully consider which feature table you would like to visualize in Empress. You can use either:
- A filtered table that matches the ordination (e.g. with rarefaction done, and/or with low-abundance features/samples removed), or
- A raw table -- that is, the original table before performing rarefaction/filtering for the ordination.
There are some pros and cons for either of these choices. If you use a filtered table, then the Empress visualization will include less data than in the raw dataset: this will impact sample presence information, sample metadata coloring, and other parts of the visualization. If you select the raw table, you might find that some nodes in the tree won't be represented by any of the samples in the ordination (if the ordination was made using a filtered table, and --p-no-shear-to-table is used). If you'd like to read more about this, there's some informal discussion in pull request 237.
The commands in this README use the raw dataset. The Empire plot command removes extra samples not represented in the ordination using the --p-filter-extra-samples flag.
Publication and Citation
An open-access publication describing Empress is available in mSystems here. If you use Empress in your work, please cite it! The BibTeX for this publication is:
@article {CantrellFedarko2021empress,
author = {Cantrell, Kalen and Fedarko, Marcus W. and Rahman, Gibraan and McDonald, Daniel and Yang, Yimeng and Zaw, Thant and Gonzalez, Antonio and Janssen, Stefan and Estaki, Mehrbod and Haiminen, Niina and Beck, Kristen L. and Zhu, Qiyun and Sayyari, Erfan and Morton, James T. and Armstrong, George and Tripathi, Anupriya and Gauglitz, Julia M. and Marotz, Clarisse and Matteson, Nathaniel L. and Martino, Cameron and Sanders, Jon G. and Carrieri, Anna Paola and Song, Se Jin and Swafford, Austin D. and Dorrestein, Pieter C. and Andersen, Kristian G. and Parida, Laxmi and Kim, Ho-Cheol and V{\'a}zquez-Baeza, Yoshiki and Knight, Rob},
editor = {Hug, Laura A.},
title = {EMPress Enables Tree-Guided, Interactive, and Exploratory Analyses of Multi-omic Data Sets},
volume = {6},
number = {2},
elocation-id = {e01216-20},
year = {2021},
doi = {10.1128/mSystems.01216-20},
publisher = {American Society for Microbiology Journals},
URL = {https://msystems.asm.org/content/6/2/e01216-20},
eprint = {https://msystems.asm.org/content/6/2/e01216-20.full.pdf},
journal = {mSystems}
}
Acknowledgements
This work is supported by IBM Research AI through the AI Horizons Network. For more information visit the IBM AI Horizons Network website.
Empress' JavaScript code is distributed with the source code of various
third-party dependencies (in the empress/support_files/vendor/ directory).
Please see
DEPENDENCY_LICENSES.md
for copies of these dependencies' licenses.


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file empress-1.2.0.tar.gz.
File metadata
- Download URL: empress-1.2.0.tar.gz
- Upload date:
- Size: 302.7 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.4.2 importlib_metadata/4.6.1 pkginfo/1.7.1 requests/2.26.0 requests-toolbelt/0.9.1 tqdm/4.61.2 CPython/3.7.10
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
73582a78746d35b9fbe062e67a79f5626faf02bb884125f9aca2df0b50087e38
|
|
| MD5 |
16b294d60bbba29769f926474a96a103
|
|
| BLAKE2b-256 |
6613a83b7939de11f10252033ea4b65db88aae5e249e5411364ea116c91d8673
|







