ML model for predicting ChIP-seq peaks in new cell types from ENCODE cell lines

Project description




Epitome
Pipeline for predicting ChIP-seq peaks in novel cell types using chromatin accessibility.
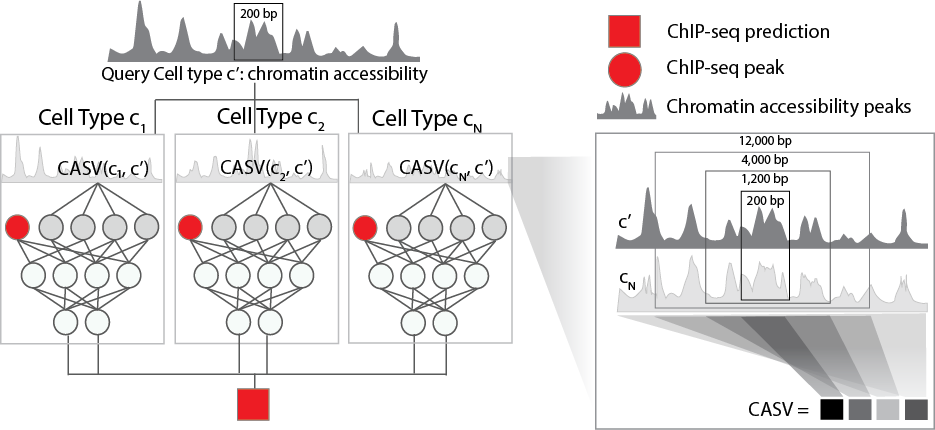
Epitome leverages chromatin accessibility (either DNase-seq or ATAC-seq) to predict epigenetic events in a novel cell type of interest. Such epigenetic events include transcription factor binding sites and histone modifications. Epitome computes chromatin accessibility similarity between ENCODE cell types and the novel cell type, and uses this information to transfer known epigentic signal to the novel cell type of interest.
Documentation
Epitome documentation is hosted at readthedocs. Documentation for Epitome includes tutorials for creating Epitome datasets, training, testing, and evaluated models.
Requirements
- conda
- python >= 3.6
Setup and Installation
- Create and activate a conda environment:
conda create --name EpitomeEnv python=3.6 pip
source activate EpitomeEnv
- Install Epitome:
pip install epitome
Training a Model
First, create an Epitome dataset that defines the cell types and ChIP-seq targets you want to train on,
from epitome.dataset import *
targets = ['CTCF','RAD21','SMC3']
celltypes = ['K562', 'A549', 'GM12878']
dataset = EpitomeDataset(targets=targets, cells=celltypes)
Now, you can create and train your model:
from epitome.models import *
model = EpitomeModel(dataset, test_celltypes = ["K562"])
model.train(5000) # train for 5000 batches
Evaluate a Model:
model.test(1000) # evaluate how well the model performs on a validation chromosome
Using Epitome on your own dataset:
Epitome can perform genome wide predictions or region specific predictions on a sample that has either DNase-seq or ATAC-seq.
To score specific regions:
chromatin_peak_file = ... # path to peak called ATAC-seq or DNase-seq in bed format
regions_file = ... # path to bed file of regions to score
results = model.score_peak_file([chromatin_peak_file], regions_file)
To score on the whole genome:
chromatin_peak_file = ... # path to peak called ATAC-seq or DNase-seq in bed format
file_prefix = ... # file to save compressed numpy predictions to.
model.score_whole_genome([chromatin_peak_file], file_prefix)
Install Epitome for development
To build Epitome for development, run:
make develop
Running unit tests
make test
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file epitome-0.0.1.tar.gz.
File metadata
- Download URL: epitome-0.0.1.tar.gz
- Upload date:
- Size: 40.7 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.4.2 importlib_metadata/4.8.1 pkginfo/1.7.1 requests/2.26.0 requests-toolbelt/0.9.1 tqdm/4.62.0 CPython/3.9.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
b260b120074abe240fc9710520ae9ceadc99696550825428a7f6b3617835fb28
|
|
| MD5 |
d5767071d71ca2b50d12d69a99cae11c
|
|
| BLAKE2b-256 |
fc3f18e18c72ff5d29dcaddc96f7437bab30bb8288157fccfe0d47c3b6f37f48
|
File details
Details for the file epitome-0.0.1-py3.9.egg.
File metadata
- Download URL: epitome-0.0.1-py3.9.egg
- Upload date:
- Size: 97.6 kB
- Tags: Egg
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.4.2 importlib_metadata/4.8.1 pkginfo/1.7.1 requests/2.26.0 requests-toolbelt/0.9.1 tqdm/4.62.0 CPython/3.9.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
18be680d3f7b2bbb0faeebc5b52d3038940b3310099c6f1b9dd05ba967cc3827
|
|
| MD5 |
efdebd8b52ae7ed00b5221f37652720f
|
|
| BLAKE2b-256 |
f650d43031a6691a687829372133819ad5ac07c7a524884497cfa128814c3d4d
|







