Quality control tools for NGS alignment file


Project description
“ezBAMQC, a tool to check the quality of mapped next generation sequencing files.”
- Codeology Icon:
-
- Description:
ezBAMQC is a tool to check the quality of either one or many mapped next-generation-sequencing datasets. It conducts comprehensive evaluations of aligned sequencing data from multiple aspects including: clipping profile, mapping quality distribution, mapped read length distribution, genomic/transcriptomic mapping distribution, inner distance distribution (for paired-end reads), ribosomal RNA contamination, transcript 5’ and 3’ end bias, transcription dropout rate, sample correlations, sample reproducibility, sample variations. It outputs a set of tables and plots and one HTML page that contains a summary of the results. Many metrics are designed for RNA-seq data specifically, but ezBAMQC can be applied to any mapped sequencing dataset such as RNA-seq, CLIP-seq, GRO-seq, ChIP-seq, DNA-seq and so on.
- Links:
- Authors:
Ying Jin, David Molik, and Molly Hammell
- Version:
0.6.7
- Contact:
Ying Jin (yjin@cshl.edu)
Installation guide for ezBAMQC for from source installs
When installing ezBAMQC there are several options, but the main point is: since ezBAMQC uses C++ STD 11 you’ll need a version of GCC that can support that, this useally means 4.8 or 4.9. beyond that, you’ll need Python, R and Corrplot for interfacing with the C code.
- Intallation:
- Prerequisites:
GCC 4.8.1 or greater GCC 4.9.1 or greater is recomended for PyPi install
- Notes:
While there are multiple methods of installing the prerequistes it may help to look at (if using a yum based linux distro):*
Devtoolset-3 for GCC compilers
IUS for Python2.7
Software Collections for collections of software (like devtoolset 3 or python)
rpmfinder for searching rpms across mutliple systems
Setup
Make sure that the GCC comiler is in your PATH:
export PATH=/path/to/gcc:$PATH
Make sure that python2.7 is in your PYTHONPATH:
export PYTHONPATH=/path/to/python2.7/site-packages:$PYTHONPATH
There are three methods of installation of ezBAMQC, from source, from setup.py, and from pypi, once prequistes are setup.
From Source
Download source
Unpack tarball and go to the directory of the package:
tar xvfz bamqc-0.6.7.tar.gz cd bamqc-0.6.7
Run make:
make
From Setup.py
python2.7 setup.py install
From Pypi
pip2.7 install BAMqc
Usage
ezBAMQC [-h] -i alignment_files [alignment_files ...] -r [refgene] [-f [attrID]] [--rRNA [rRNA]] -o [dir] [--stranded [stranded]] [-q [mapq]] [-l labels [labels ...]] [-t NUMTHREADS]
optional arguments:
-h, --help show this help message and exit.
-i, --inputFile alignment files. Could be multiple SAM/BAM files separated by space. Required.
-r, --refgene gene annotation file in GTF format. Required
-f the read summation at which feature level in the GTF file. DEFAULT: gene_id.
--rRNA rRNA coordinates in BED format.
-o, --outputDir output directory. Required.
--stranded strandness of the library?
yes : sense stranded
reverse : reverse stranded
no : not stranded
DEFAULT: yes.
-q, --mapq Minimum mapping quality (phred scaled) for an alignment to be called uniquely mapped. DEFAULT:30
-l, --label Labels of input files. DEFAULT:smp1 smp2 ...
-t, --threads Number of threads to use. DEFAULT:1
Example:
ezBAMQC -i test-data/exp_data/treat1.bam test-data/exp_data/treat2.bam test-data/exp_data/treat3.bam -r test-data/exp_data/hg9_refGene.gtf -q 30 --rRNA test-data/exp_data/hg19_rRNA.bed -o exp_output2 Please find the example output from folder test-data.
Demo
Want to try ezBAMQC before trying it? you can now utilize our tool demo hosted on our Yabi Demo to do so.
To login use username and password: |
|---|
|
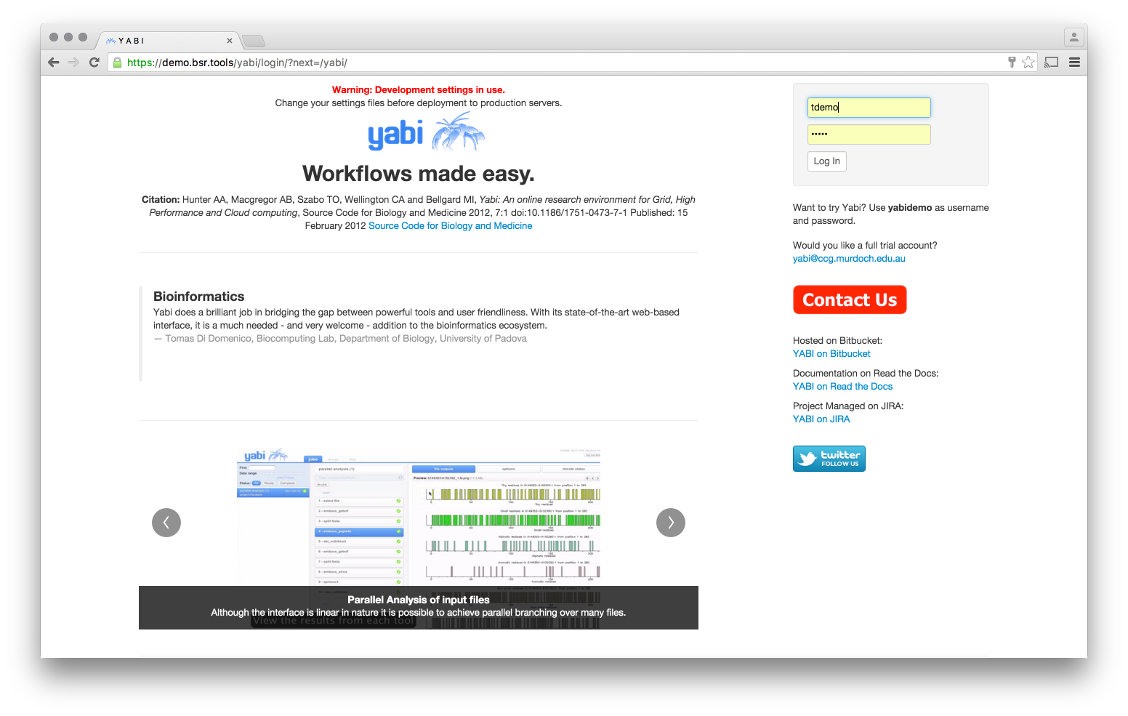
The login screen, usernname and password go in the top right corner.
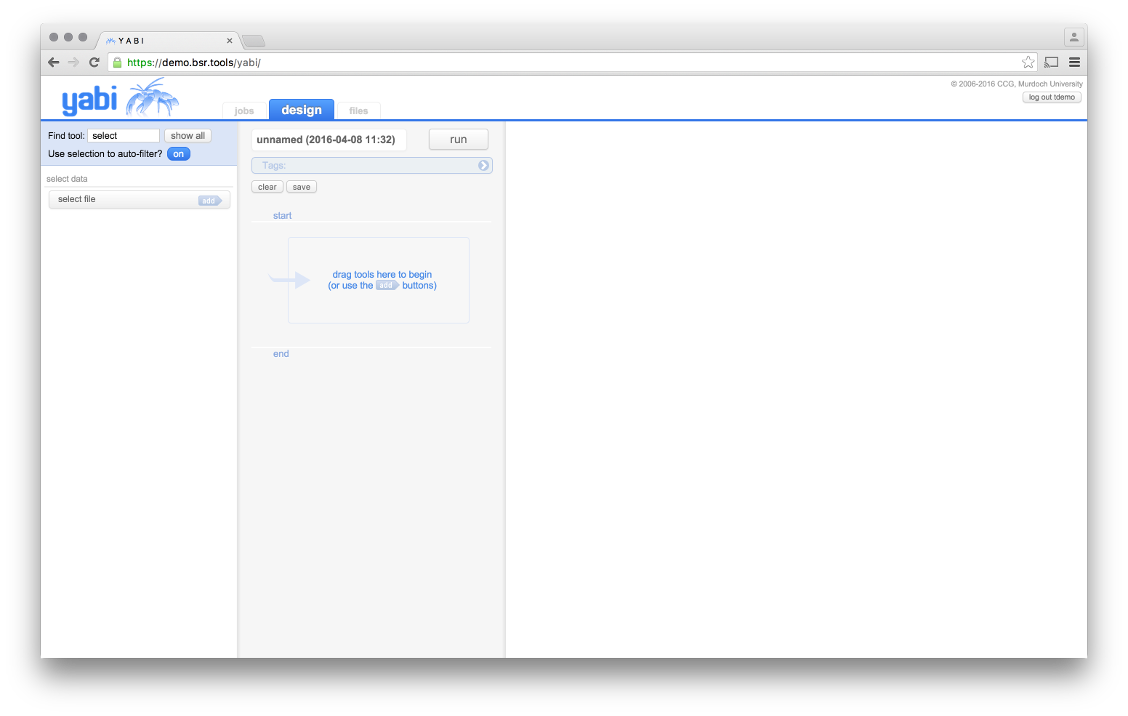
The “Design” Frame, use the “show all” button to make visable the ezBAMQC tool.
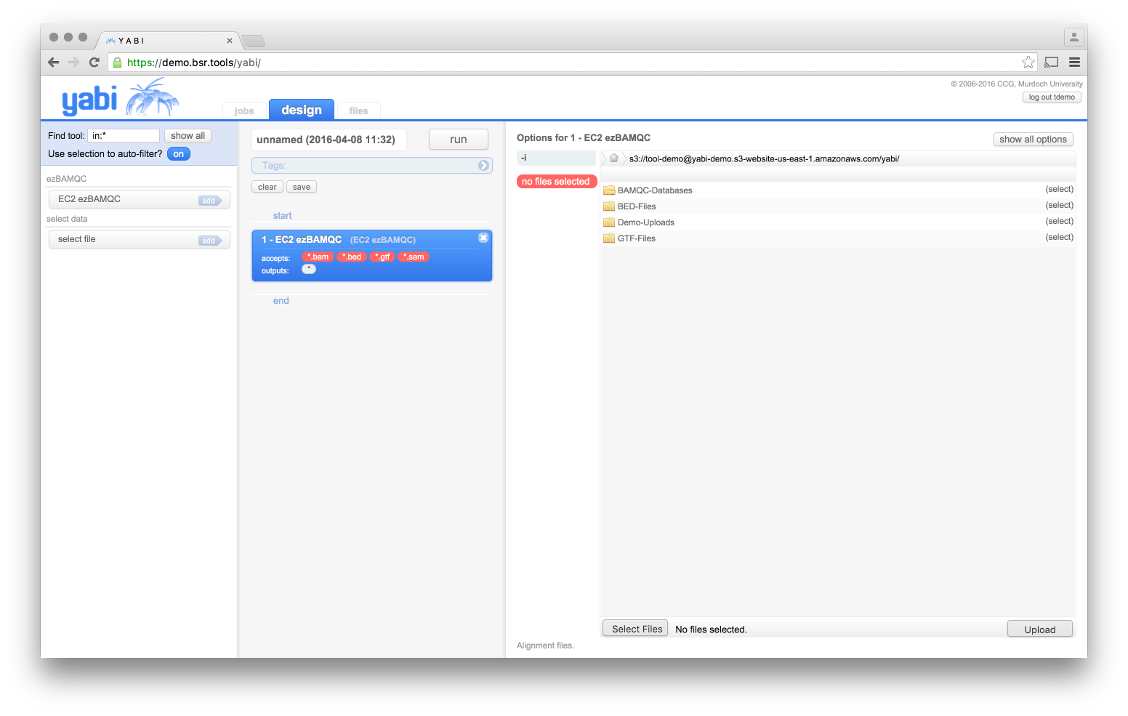
The ezBAMQC tool page, select appropriate files from the S3 instance or upload your own.
A note on Yabi, Yabi was created by the Centre For Comparative Genomics, https://ccg.murdoch.edu.au/ . You can check our their more extensive Yabi Demo, https://ccgapps.com.au/yabi/ or their Yabi Wiki, https://bitbucket.org/ccgmurdoch/yabi/wiki/Home for more information.
FAQ
Q: Why use ezBAMQC?
A: ezBAMQC is efficient and easy to use. With one command line, it reports a comprehensive evaluation of the data with a set of plots and tables.The ability to assess multiple samples together with high efficiency make it especially useful in cases where there are a large number of samples from the same condition, genotype, or treatment. ezBAMQC was written in C++ and supports multithreading. A mouse RNA-seq sample with 120M alignments can be done in 8 minutes with 5 threads.
Q: Why the total number of reads reported by ezBAMQC does not match with samtools flagstat?
A: The difference is because of non-uniquely mapped reads or multiply aligned reads (multi-reads). Samtools flagstat counts each multiple aligment as a different reads, but ezBAMQC counts reads accoriding to the read ID, i.e., each individual read will be counted once no matter that it is a uniquely mapped read or multi-read.
Q: What is “Low Quality Reads” ?
A: Reads marked as qc fail accoriding to SAM format or reads with mapping quality lower than the value set by the option -q will be considered as “Low Quality Reads”.
Q: How the setting of option -q alter the results?
A: Reads with low quality, i.e., did not pass -q cutoff, are only counted in Total Reads, Mapped Reads, and Mappability by mapping quality plot. The rest of the report does not include low quality reads.
Q: Do multi-reads (non-uniquely mapped reads) have been considered in Read distribution and gene quantification?
A: No. Only uniquely mapped reads were counted.
Acknowledgements
Samtools contributors
Users’ valuable feedback
Copying & Distribution
ezBAMQC is free software: you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation, either version 3 of the License, or (at your option) any later version.
This program is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU General Public License for more details.
You should have received a copy of the GNU General Public License along with ezBAMQC. If not, see this website
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file ezBAMQC-0.6.7.tar.gz.
File metadata
- Download URL: ezBAMQC-0.6.7.tar.gz
- Upload date:
- Size: 2.1 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
478560c3763bbca11e1dba3311eae83968fdcb7b8f0279801032466cf2cb7fed
|
|
| MD5 |
555b8b2911372b17d9f50474a86b44db
|
|
| BLAKE2b-256 |
ebebea7db33f3d7cac11e837e82ddc4f238947b855a7ae99ec1e6d2206b1a070
|







