A Nextflow pipeline assembler for genomics. Pick your modules. Assemble them. Run the pipeline.


Project description
FlowCraft :whale2::package:









A Nextflow pipeline assembler for genomics. Pick your modules. Assemble them. Run the pipeline.
(Previously known as Assemblerflow)
The premisse
Build a pipeline
What if building your own genomics pipeline would be as simple as:
flowcraft.py build -t "trimmomatic fastqc skesa pilon" -o my_pipeline.nf
Seems pretty simple right? What if we could run this pipeline with a single command on any linux machine or cluster by leveraging the awesomeness of nextflow and docker/singularity containers without having to install any of the pipeline dependencies?
Run the pipeline
nextflow run my_pipeline.nf --fastq path/to/fastq
N E X T F L O W ~ version 0.30.1
Launching `my_pipeline.nf` [admiring_lamarck] - revision: 82cc9cd2ed
============================================================
M Y P I P E L I N E
============================================================
Built using flowcraft v1.2.1
Input FastQ : 2
Input samples : 1
Reports are found in : ./reports
Results are found in : ./results
Profile : standard
Starting pipeline at Tue Jun 12 19:38:26 WEST 2018
[warm up] executor > local
[7c/eb5f2f] Submitted process > integrity_coverage_1_1 (02AR0553)
(...)
[31/7d90a1] Submitted process > compile_pilon_report_1_6
Completed at: Tue Jun 12 19:58:32 WEST 2018
Duration : 20m 6s
Success : true
Exit status : 0
Congratulations! You just built and executed your own pipeline with only two commands! :tada:
Installation
FlowCraft is available as a bioconda package, which already brings nextflow:
conda install flowcraft
Container engines
Pipelines built with FlowCraft require at least one container
engine to be installed, among docker, singularity or shifter.
If you already have any one of these installed, you're good to go.
If not, we recommend installing singularity, though it should be installed with
root privileges and accessible in all compute nodes.
How to use it
The complete user guide of FlowCraft can be found on readthedocs.org. For a quick and dirty demonstration, see below.
Quick guide
Building a pipeline
FlowCraft comes with a number of ready-to-use components to build your
own pipeline. Following some basic rules, such as the output type of one process
must match the input type of the next process, assembling a pipeline is done
using the build mode and the -t option:
flowcraft build -t "trimmomatic spades abricate" -o my_pipeline.nf -n "assembly pipe"
This command will generate everything that is necessary to run the
pipeline automatically, but the main pipeline executable
file will be my_pipeline.nf. This file will contain a nextflow pipeline
for genome assembly starts with trimmomatic and finishes with anti-microbial
gene annotation using abricate.
Wait... what about the software parameters?
Each component in the pipeline has its own set of parameters that can be
modified before or when executing the pipeline. These parameters are
described in the documentation of each process and you can check the options
of your particular pipeline using the help option:
$ nextflow run my_pipeline.nf --help
N E X T F L O W ~ version 0.30.1
Launching `my_pipeline.nf` [prickly_picasso] - revision: 2e1a226e6d
============================================================
F L O W C R A F T
============================================================
Built using flowcraft v1.2.1
Usage:
nextflow run my_pipeline.nf
--fastq Path expression to paired-end fastq files. (default: fastq/*_{1,2}.*) (default: 'fastq/*_{1,2}.*')
Component 'INTEGRITY_COVERAGE_1_1'
----------------------------------
--genomeSize_1_1 Genome size estimate for the samples in Mb. It is used to estimate the coverage and other assembly parameters andchecks (default: 1)
--minCoverage_1_1 Minimum coverage for a sample to proceed. By default it's setto 0 to allow any coverage (default: 0)
Component 'TRIMMOMATIC_1_2'
---------------------------
--adapters_1_2 Path to adapters files, if any. (default: 'None')
--trimSlidingWindow_1_2 Perform sliding window trimming, cutting once the average quality within the window falls below a threshold (default: '5:20')
--trimLeading_1_2 Cut bases off the start of a read, if below a threshold quality (default: 3)
--trimTrailing_1_2 Cut bases of the end of a read, if below a threshold quality (default: 3)
--trimMinLength_1_2 Drop the read if it is below a specified length (default: 55)
Component 'FASTQC_1_3'
----------------------
--adapters_1_3 Path to adapters files, if any. (default: 'None')
Component 'ASSEMBLY_MAPPING_1_5'
--------------------------------
--minAssemblyCoverage_1_5 In auto, the default minimum coverage for each assembled contig is 1/3 of the assembly mean coverage or 10x, if the mean coverage is below 10x (default: 'auto')
--AMaxContigs_1_5 A warning is issued if the number of contigs is overthis threshold. (default: 100)
--genomeSize_1_5 Genome size estimate for the samples. It is used to check the ratio of contig number per genome MB (default: 2.1)
This help message is dynamically generated depending on the pipeline you build.
Since this pipeline starts with trimmomatic, which receives fastq files as input,
--fastq is the default parameter for providing paired-end fastq files.
Running a pipeline
Now that we have our nextflow pipeline built, we are ready to executed it by
providing input data. By default, FlowCraft pipelines will run locally and use
singularity to run the containers of each component. This can be
changed in multiple ways, but for convenience FlowCraft has already defined
profiles for most configurations of executors and container engines.
Running a pipeline locally with singularity can be done with:
# Pattern for paired-end fastq is '<sample>_1.fastq.gz <sample>_2.fastq.gz'
nextflow run my_pipeline --fastq "path/to/fastq/*_{1,2}.*"
If you want to run a pipeline in a cluster with SLURM and singularity, just use the appropriate profile:
nextflow run my_pipeline --fastq "path/to/fastq/*_{1,2}.*" -profile slurm_sing
During the execution of the pipeline, the results and reports for each component
are continuously saved to the results and reports directory, respectively.
Inspecting a pipeline progress
Since version 1.2.0, it is possible to inspect the progress of a nextflow pipeline
using the flowcraft inspect mode. To check the progress in a terminal, simply
type:
flowcraft inspect
On the directory where the pipeline is running. Alternatively, you can view the progress
in FlowCraft's web service by using the broadcast option:
flowcraft inspect -m broadcast
Why not just write a Nextflow pipeline?
In many cases, building a static nextflow pipeline is sufficient for our goals. However, when building our own pipelines, we often felt the need to add dynamism to this process, particularly if we take into account how fast new tools arise and existing ones change. Our biological goals also change over time and we might need different pipelines to answer different questions. FlowCraft makes this very easy, by having a set of pre-made and ready-to-use components that can be freely assembled.
For instance, changing the assembly software in a genome assembly pipeline becomes as easy as:
# Use spades
trimmomatic spades pilon
# Use skesa
trimmomatic skesa pilon

If you are interested in having some sort of genome annotation, simply add those components at the end, using a fork syntax:
# Run prokka and abricate at the end of the assembly
trimmomatic spades pilon (prokka | abricate)
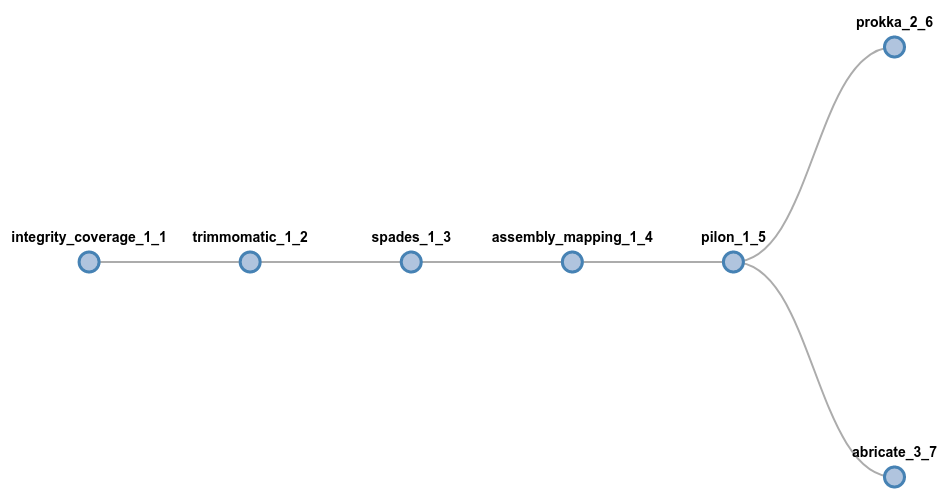
On the other hand, if you are interest in just perform allele calling for wgMLST,
simply add chewbbaca:
trimmomatic spades pilon chewbbaca

Since nextflow handles parallelism of large sets of data so well, simple pipelines of two components are also useful to build:
trimmomatic fastqc
As the number of existing components grow, so does your freedom to build pipelines.
Roadmap
You can see what we're planning next on our roadmap guide.
Developer guide
Adding new components
Is there a missing component that you would like to see included? We would love to expand! You could make a component request in our issue tracker.
If you want to be part of the team, you can contribute with the code as well. Each component in FlowCraft can be independently added without having to worry about the rest of the code base. You'll just need to have some knowledge of python and nextflow. Check the developer documentation for how-to guides
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file flowcraft-1.4.1.tar.gz.
File metadata
- Download URL: flowcraft-1.4.1.tar.gz
- Upload date:
- Size: 1.4 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/1.13.0 pkginfo/1.4.2 requests/2.19.1 setuptools/39.1.0 requests-toolbelt/0.9.1 tqdm/4.31.1 CPython/3.6.8
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
72613803632a73c60beb6b399c9aa9eb40cfa201df59879a3e50a2bddc65b04e
|
|
| MD5 |
8e863d544701fcf719059da4d78cf479
|
|
| BLAKE2b-256 |
7de0c68b357b4d3ff14df66a69669ecb1a1389f7dd0c77800706f43981d36b0c
|
File details
Details for the file flowcraft-1.4.1-py3-none-any.whl.
File metadata
- Download URL: flowcraft-1.4.1-py3-none-any.whl
- Upload date:
- Size: 1.5 MB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/1.13.0 pkginfo/1.4.2 requests/2.19.1 setuptools/39.1.0 requests-toolbelt/0.9.1 tqdm/4.31.1 CPython/3.6.8
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
b21b922357e37fbfecae9c91cb700153e4402c5d7195b935182f0ca94a3a5c81
|
|
| MD5 |
a4c0da8e9372cc924b7bdac61c9babfe
|
|
| BLAKE2b-256 |
a5e9f6ba22f315e2819962cad2be4c76feafe8e350bbe49bed02a0a646a410a8
|







