A package for detecting epsitasis by machine learning

Project description
GenEpi
GenEpi is a package to uncover epistasis associated with phenotypes by a machine learning approach, developed by Yu-Chuan Chang at c4Lab of National Taiwan University and Taiwan AI Labs
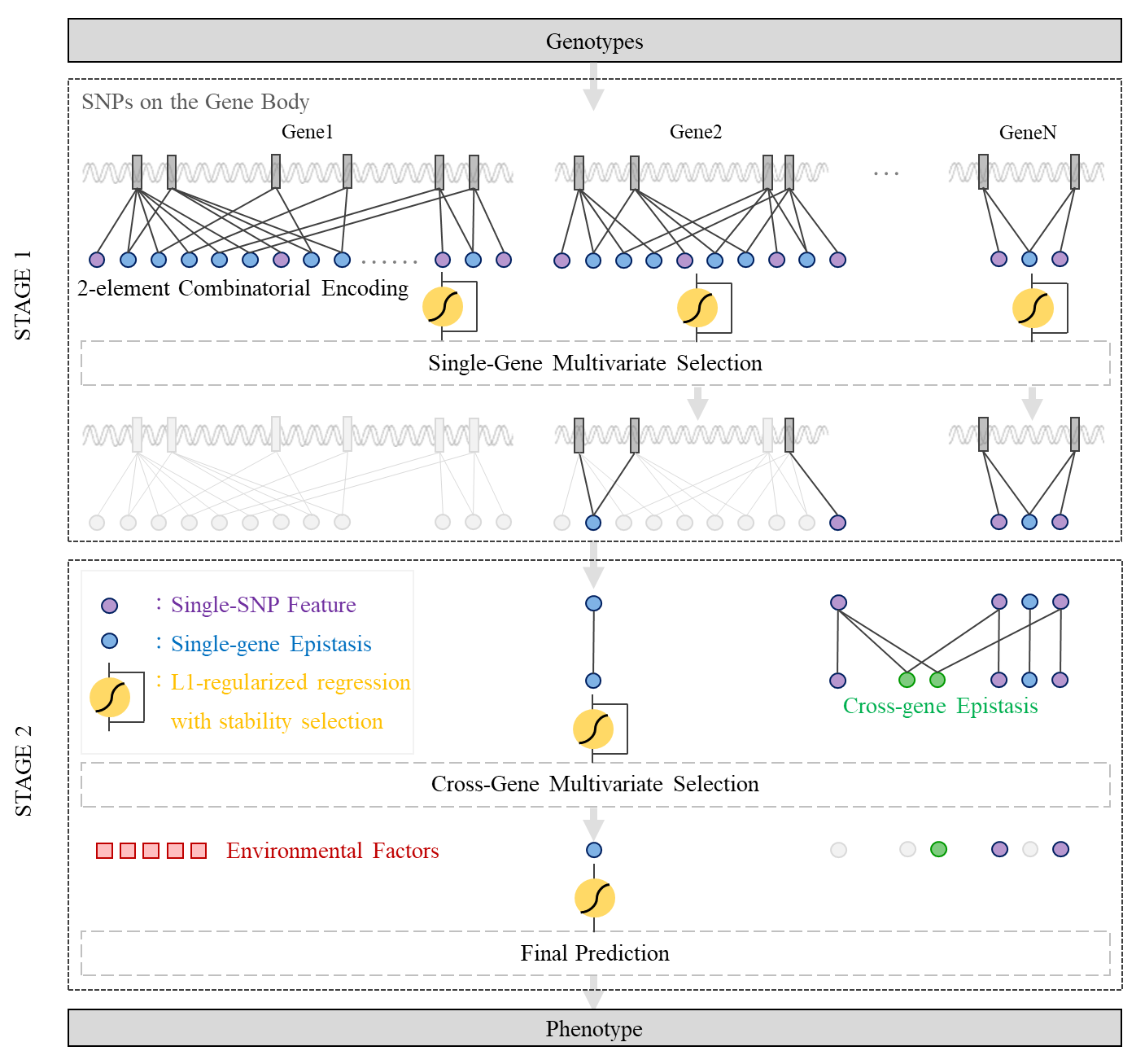
The architecture and modules of GenEpi.
Introduction
GenEpi is designed to group SNPs by a set of loci in the gnome. For examples, a locus could be a gene. In other words, we use gene boundaries to group SNPs. A locus can be generalized to any particular regions in the genome, e.g. promoters, enhancers, etc. GenEpi first considers the genetic variants within a particular region as features in the first stage, because it is believed that SNPs within a functional region might have a higher chance to interact with each other and to influence molecular functions.
GenEpi adopts two-element combinatorial encoding when producing features and models them by L1-regularized regression with stability selection In the first stage (STAGE 1) of GenEpi, the genotype features from each single gene will be combinatorically encoded and modeled independently by L1-regularized regression with stability selection. In this way, we can estimate the prediction performance of each gene and detect within-gene epistasis with a low false positive rate. In the second stage (STAGE 2), both of the individual SNP and the within-gene epistasis features selected by STAGE 1 are pooled together to generate cross-gene epistasis features, and modeled again by L1-regularized regression with stability selection as STAGE 1. Finally, the user can combine the selected genetic features with environmental factors such as clinical features to build the final prediction models.
Standalone App
(Latest Update!) The standalone and installation free app - AppGenEpi (v.beta) is now released. Just download it and have fun.
| OS | Version | Link |
|---|---|---|
| MacOS | Catalina | AppGenEpi_MacOS_beta |
| Linux | CentOS 7 | AppGenEpi_Linux_beta |
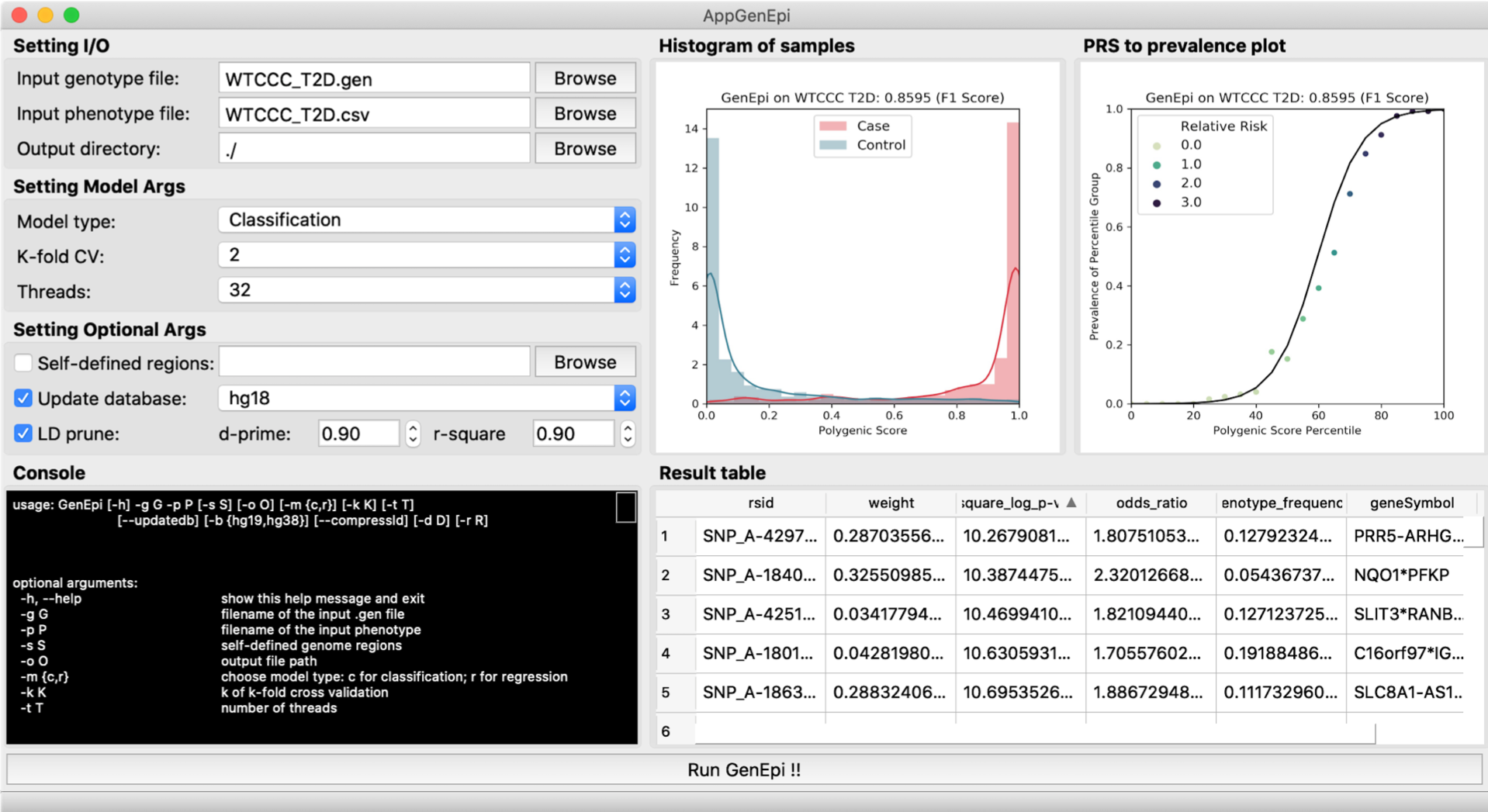
The snapshot of AppGenEpi.
For MacOS
.1) unzip AppGenEpi_MacOS_beta.zip; 2) drag AppGenEpi.app to Applications; 3) allow permission for running AppGenEpi.app by setting System Preferences > Security & Privacy (We are not identified developers so far.).
For Linux
.1) change the directory to AppGenEpi; 2) use ./AppGenEpi to run it.
Citing
Please considering cite the following paper if you use GenEpi in a scientific publication:
[1] Yu-Chuan Chang, June-Tai Wu, Ming-Yi Hong, Yi-An Tung, Ping-Han Hsieh, Sook Wah Yee, Kathleen M. Giacomini, Yen-Jen Oyang, and Chien-Yu Chen. "Genepi: Gene-Based Epistasis Discovery Using Machine Learning." BMC Bioinformatics 21, 68 (2020). https://doi.org/10.1186/s12859-020-3368-2
Quickstart
This section gets you started quickly. The completed GenEpi's documentation please find on Welcome to GenEpi’s docs!
Installation
$ pip install GenEpi
NOTE: GenEpi is a memory-consuming package, which might cause memory errors when calculating the epistasis of a gene containing a large number of SNPs. We recommend that the memory for running GenEpi should be over 256 GB.
Running a quick test
Please use following command to run a quick test, you will obtain all the outputs of GenEpi in your current folder.
$ GenEpi -g example -p example -o ./
Interpreting the main results table
GenEpi will automatically generate three folders (snpSubsets, singleGeneResult, crossGeneResult) beside your .GEN file. You could go to the folder crossGeneResult directly to obtain your main table for episatasis in Result.csv.
| RSID | -Log10(χ2 p-value) | Odds Ratio | Genotype Frequency | Gene Symbol |
|---|---|---|---|---|
| rs157580_BB rs2238681_AA | 8.4002 | 9.3952 | 0.1044 | TOMM40 |
| rs449647_AA rs769449_AB | 8.0278 | 5.0877 | 0.2692 | APOE |
| rs59007384_BB rs11668327_AA | 8.0158 | 12.0408 | 0.0824 | TOMM40 |
| rs283811_BB rs7254892_AA | 8.0158 | 12.0408 | 0.0824 | PVRL2 |
| rs429358_AA | 5.7628 | 0.1743 | 0.5962 | APOE |
| rs73052335_AA rs429358_AA | 5.6548 | 0.1867 | 0.5714 | APOC1*APOE |
The first column lists each feature by its RSID and the genotype (denoted as RSID_genotype), the pairwise epistatis features are represented using two SNPs. The last column describes the genes where the SNPs are located according to the genomic coordinates. We used a star sign to denote the epistasis between genes. The p-values of the χ2 test (the quantitative task will use student t-test) are also included. The odds ratio significantly away from 1 also indicates whether the features are potential causal or protective genotypes. Since low genotype frequency may cause unreliable odds ratios, we also listed this information in the table.
Options
For checking all the optional arguments, please use --help:
$ GenEpi --help
You will obtain the following argument list:
usage: GenEpi [-h] -g G -p P [-s S] [-o O] [-m {c,r}] [-k K] [-t T]
[--updatedb] [-b {hg19,hg38}] [--compressld] [-d D] [-r R]
optional arguments:
-h, --help show this help message and exit
-g G filename of the input .gen file
-p P filename of the input phenotype
-s S self-defined genome regions
-o O output file path
-m {c,r} choose model type: c for classification; r for regression
-k K k of k-fold cross validation
-t T number of threads
update UCSC database:
--updatedb enable this function
-b {hg19,hg38} human genome build
compress data by LD block:
--compressld enable this function
-d D threshold for compression: D prime
-r R threshold for compression: R square
Meta
Chester (Yu-Chuan Chang) - chester75321@gmail.com
Distributed under the MIT license. See LICENSE for more information.
https://github.com/Chester75321/GenEpi/
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file genepi-2.0.10.tar.gz.
File metadata
- Download URL: genepi-2.0.10.tar.gz
- Upload date:
- Size: 448.8 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.2.0 pkginfo/1.5.0.1 requests/2.24.0 setuptools/49.2.1 requests-toolbelt/0.9.1 tqdm/4.48.2 CPython/3.7.8
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
c093f7642a9cd1b49914ac5532e797e0ac6e3cf43d0b71f7feee84d4406b4c18
|
|
| MD5 |
fc7e875c3c45bd06ca9063f3a701ec11
|
|
| BLAKE2b-256 |
785ac4e7d26a49ebae872df7d898e13eab1d8d668f0f28bc699fcfd268ea568c
|
File details
Details for the file genepi-2.0.10-py3-none-any.whl.
File metadata
- Download URL: genepi-2.0.10-py3-none-any.whl
- Upload date:
- Size: 470.4 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.2.0 pkginfo/1.5.0.1 requests/2.24.0 setuptools/49.2.1 requests-toolbelt/0.9.1 tqdm/4.48.2 CPython/3.7.8
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
cdde22637c961aaaf734ff9a66ff074b24e39d923426b705561591b03f314660
|
|
| MD5 |
b109f1198244ba0f783b83c69da7c41f
|
|
| BLAKE2b-256 |
f4c5da57e134815e7d3587924f73c80a030097908af52ecb86cc58bedac46d5f
|







