Python toolkit for handling GWAS summary statistics

Project description
GWASLab




- A handy Python-based toolkit for handling GWAS summary statistics (sumstats).
- Each process is modularized and can be customized to your needs.
- Sumstats-specific manipulations are designed as methods of a Python object,
gwaslab.Sumstats.
Installation
install via pip
The latest version of GWASLab now supports Python 3.9, 3.10, 3.11, and 3.12.
pip install gwaslab
install in conda environment
Create a Python 3.9, 3.10, 3.11 or 3.12 environment and install gwaslab using pip:
conda env create -n gwaslab -c conda-forge python=3.12
conda activate gwaslab
pip install gwaslab
or create a new environment using yml file environment.yml
conda env create -n gwaslab -f environment.yml
install using docker (deprecated)
A docker file is available here for building local images.
Quick start
import gwaslab as gl
# load plink2 output
mysumstats = gl.Sumstats("sumstats.txt.gz", fmt="plink2")
# or load sumstats with auto mode (auto-detecting commonly used headers)
# assuming ALT/A1 is EA, and frq is EAF
mysumstats = gl.Sumstats("sumstats.txt.gz", fmt="auto")
# or you can specify the columns:
mysumstats = gl.Sumstats("sumstats.txt.gz",
snpid="SNP",
chrom="CHR",
pos="POS",
ea="ALT",
nea="REF",
eaf="Frq",
beta="BETA",
se="SE",
p="P",
direction="Dir",
n="N",
build="19")
# manhattan and qq plot
mysumstats.plot_mqq()
...
Documentation and tutorials
Documentation and tutorials for GWASLab are avaiable at here.
Functions
Loading and Formatting
- Loading sumstats by simply specifying the software name or format name, or specifying each column name.
- Converting GWAS sumstats to specific formats:
- LDSC / MAGMA / METAL / PLINK / SAIGE / REGENIE / MR-MEGA / GWAS-SSF / FUMA / GWAS-VCF / BED...
- check available formats
- Optional filtering of variants in commonly used genomic regions: Hapmap3 SNPs / High-LD regions / MHC region
Standardization & Normalization
- Variant ID standardization
- CHR and POS notation standardization
- Variant POS and allele normalization
- Genome build : Inference and Liftover
Quality control, Value conversion & Filtering
- Statistics sanity check
- Extreme value removal
- Equivalent statistics conversion
- BETA/SE , OR/OR_95L/OR_95U
- P, Z, CHISQ, MLOG10P
- Customizable value filtering
Harmonization
- rsID assignment based on CHR, POS, and REF/ALT
- CHR POS assignment based on rsID using a reference text file
- Palindromic SNPs and indels strand inference using a reference VCF
- Check allele frequency discrepancy using a reference VCF
- Reference allele alignment using a reference genome sequence FASTA file
Visualization
- Mqq plot: Manhattan plot, QQ plot or MQQ plot (with a bunch of customizable features including auto-annotate nearest gene names)
- Miami plot: mirrored Manhattan plot
- Brisbane plot: GWAS hits density plot
- Regional plot: GWAS regional plot
- Genetic correlation heatmap: ldsc-rg genetic correlation matrix
- Scatter plot: variant effect size comparison
- Scatter plot: allele frequency comparison
- Scatter plot: trumpet plot (plot of MAF and effect size with power lines)
Visualization Examples
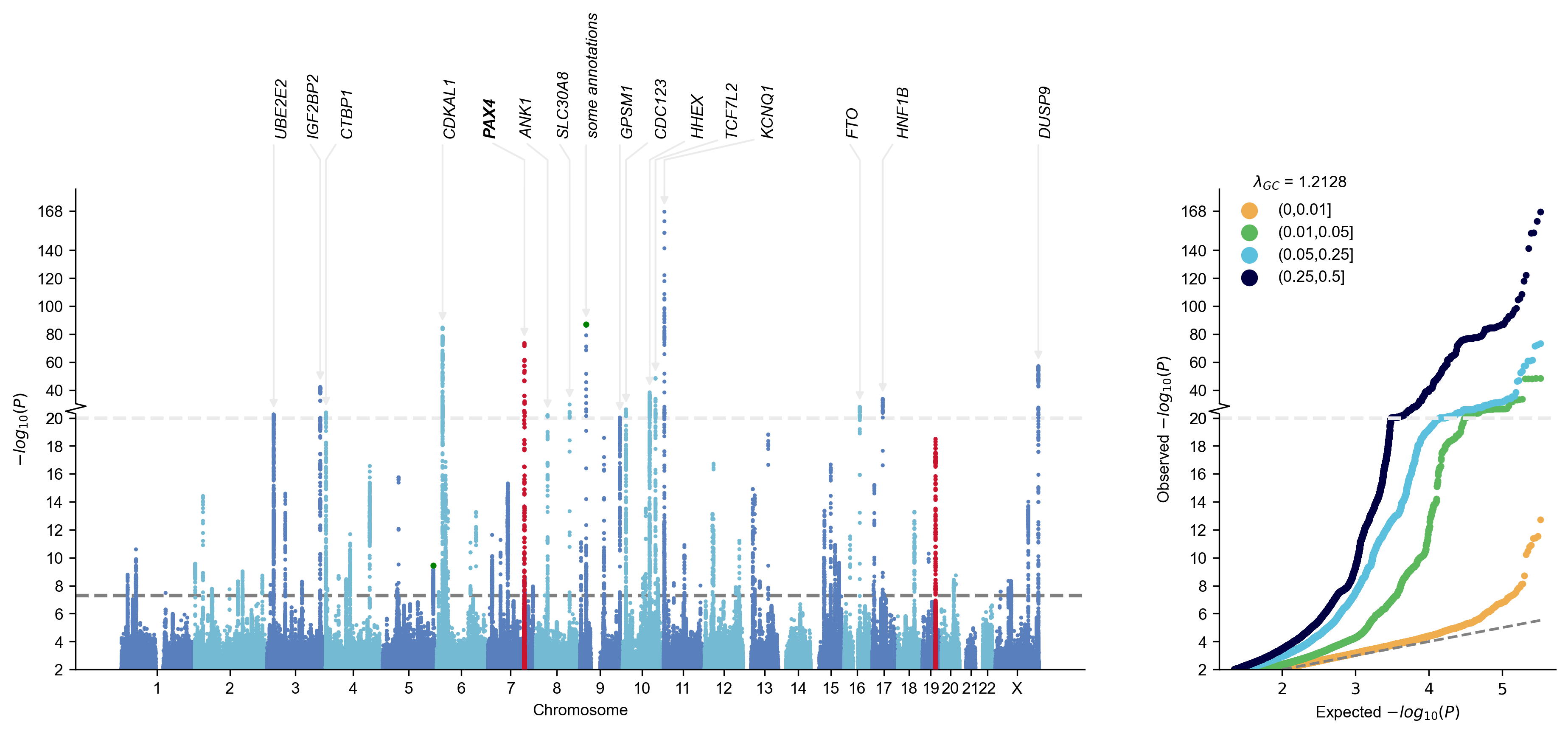
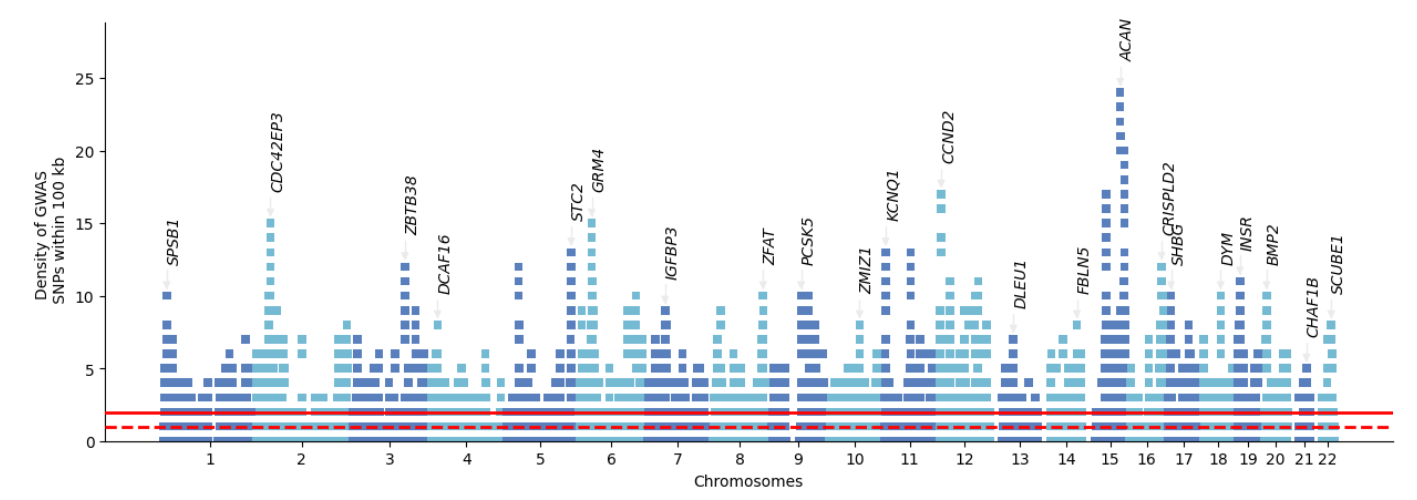
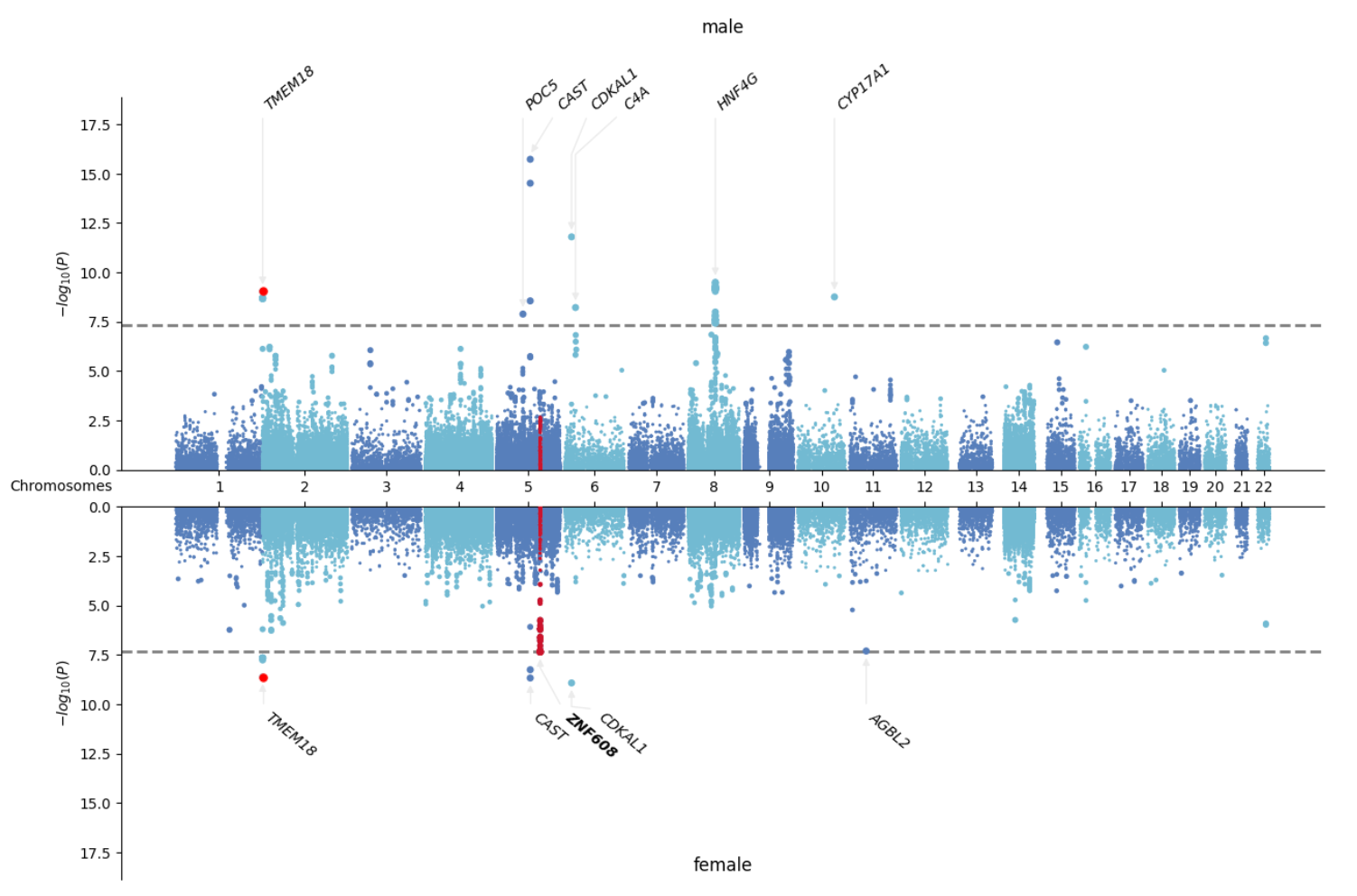
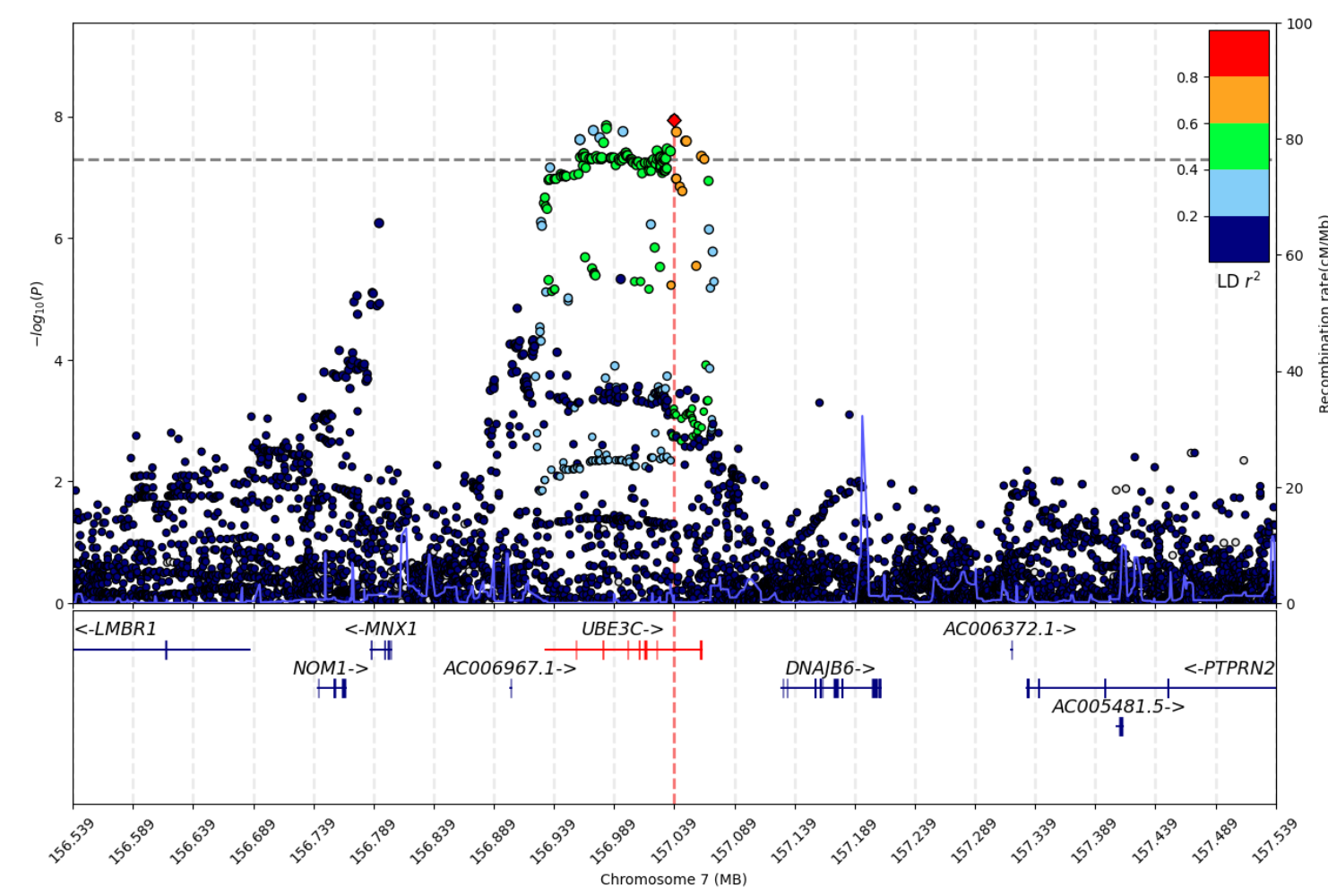
Other Utilities
- Read ldsc h2 or rg outputs directly as DataFrames (auto-parsing).
- Extract lead variants given a sliding window size.
- Extract novel loci given a list of known lead variants / or known loci obtained from GWAS Catalog.
- Logging: keep a complete record of manipulations applied to the sumstats.
- Sumstats summary: give you a quick overview of the sumstats.
- ...
Issues
- GWASLab is currently under active development, with frequent updates.
- Note: Known issues are documented at https://cloufield.github.io/gwaslab/KnownIssues/.
How to cite
- GWASLab preprint: He, Y., Koido, M., Shimmori, Y., Kamatani, Y. (2023). GWASLab: a Python package for processing and visualizing GWAS summary statistics. Preprint at Jxiv, 2023-5. https://doi.org/10.51094/jxiv.370
Sample data used for tutorial
- Sample GWAS data used in GWASLab is obtained from: http://jenger.riken.jp/ (Suzuki, Ken, et al. "Identification of 28 new susceptibility loci for type 2 diabetes in the Japanese population." Nature genetics 51.3 (2019): 379-386.).
Acknowledgement
Thanks to @sup3rgiu, @soumickmj and @gmauro for their contributions to the source codes.
Contacts
- Github: https://github.com/Cloufield/gwaslab
- Blog (in Chinese): https://gwaslab.com/
- Email: gwaslab@gmail.com
- Stats: https://pypistats.org/packages/gwaslab
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file gwaslab-4.1.3.tar.gz.
File metadata
- Download URL: gwaslab-4.1.3.tar.gz
- Upload date:
- Size: 23.0 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.12.0
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d0833aa6561804dc8aaae607e3d95d1330f2f5f17c644fe9fa320843b7fe769c
|
|
| MD5 |
6c51f5a9ad65a03db7646869f63d8e67
|
|
| BLAKE2b-256 |
cae8a17a3833b765fbff624d1eef9362852d894afa553eb8b8ca34a8ab255cc5
|
File details
Details for the file gwaslab-4.1.3-py3-none-any.whl.
File metadata
- Download URL: gwaslab-4.1.3-py3-none-any.whl
- Upload date:
- Size: 22.9 MB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.12.0
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
426ceefe3d78e439673956a0e3561826a896c370f66f55fcdc6dee3596cdf114
|
|
| MD5 |
b6bc63b8af46174085b57d85b8c4735d
|
|
| BLAKE2b-256 |
1ef2fc7e31c2d880fd76dfff8c6fab9d1d88455af63f767ceb373f195fd59dc2
|







