Automisation of graph generation for gene FC databases.

Project description
A series of scripts for gene database automation. Developed for the Philippe Campeau Laboratory.
Installation
helixPC is available on PyPI, and is easily installable through pip.
For stability, it is recommended that you install this package in a virtual environment, but you may skip this step if you do not know how to set these up.
$ pip install helixpc
That’s it! You may now call the script with:
$ helixpc
Usage
Generating a file for the graphing utility
$ helixpc group <group_input> [--nonan] [--round=[int]]
If you do not yet have a valid input file for graph generation, the command group can help you generate one automatically. Simply stick all your batches in a single csv file, call the utility and a file named output.csv will be generated. You can then feed to the graphing utility.
Please note that:
If certain genes are included multiple times, their mean will be calculated, and only a single entry will appear in the output.
You may pass [--nonan] or [-n] to omit any gene that are missing entries in a batch.
You may round each values by passing an integer to [--round] or [-r]. The integer passed is equivalent to the number of decimal places. For example, passing -r=2 will round all values to the nearest hundredth.
You may pass the name of the output file with [--output]. If not used, the output file name will default to output.csv
input file format:
Check the example group_input.csv
The first row should specify the column titles.
You must call the columns containing gene names gene_symbol, they are used as columns of reference by the scripts.
Using the graphing utility
$ helixpc graph <graph_input> [--heat] [--scatter] [--alpha] [--pvalues] <control> <sample> [<sample> ...]
Once you have a csv file that you want to use for generating graph, you may feed it to the graphing utility. You must give the csv file a series of arguments for it to function properly:
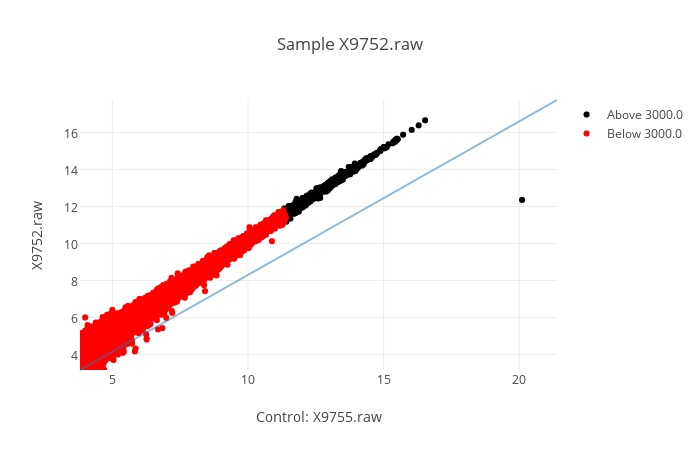
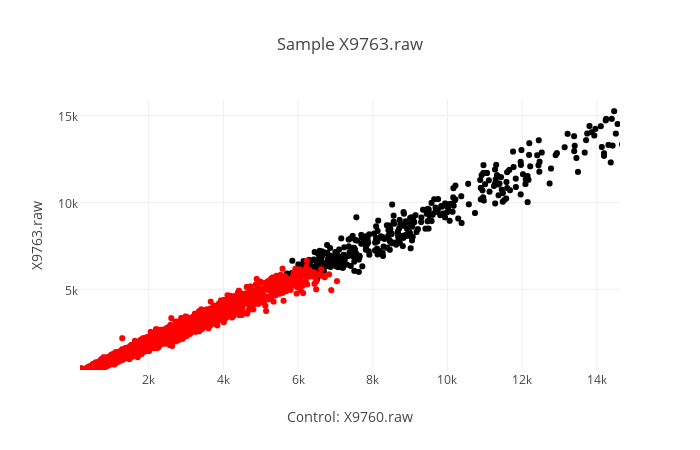
-s | --scatter
Specifies that you want scatter graph(s). Scatter graphs are generated with a control (always the same) in the x axis, and a sample in the y axis. Giving more than one sample will return to you multiple graphs, one for each sample. You can hover over each point to see the name of the gene it is representing.
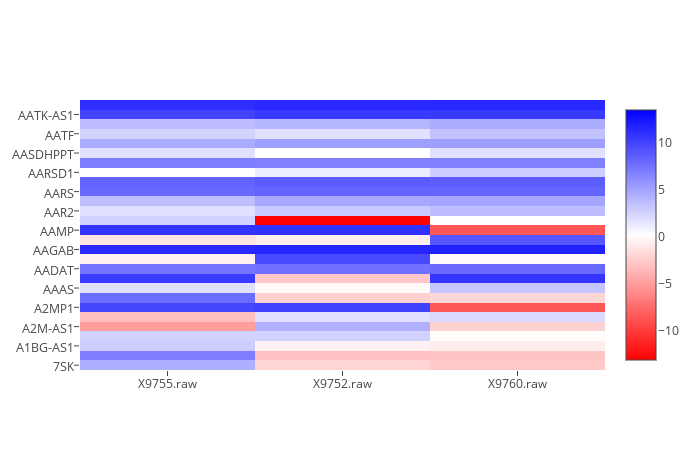
-he | --heat
Specifies that you want a heat graph. It will graph both control and samples. It accepts --no-log to prevent taking log_2() of all values. All other passed functions are ignored (for now).
<control>
Specifies the control. You may give the index or the name of a column. You may also give a series of indexes/column-names separated by a comma, and the values used will be the mean of each row for the series of columns given.
<sample>
Specifies the sample. You may give the index or the name of a column. You may also give a series of indexes/column-names separated by a comma, and the values used will be the mean of each row for the series of columns given. You may specify many samples, simply put a space in between each.
-a | --alpha
Specifies the alpha value. If used, you must also specify a column that will serve as the colour value for each gene. See --pvalues.
-p | --pvalues
Once you have specified an alpha, you must specify column(s) whose value for each gene will be compared to the alpha value. Everything below (including) the alpha value will be colored red. Everything strictly above the alpha will be colored black.
e.g. : helixpc graph output.csv -s -p=X9760.raw -a=5000 2,3 4
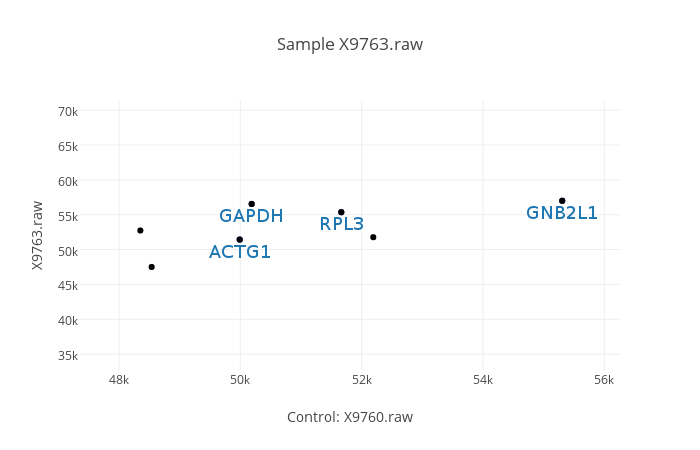
-l | --label
Specifies the column to be used for making labels. The ten lowest and ten highest values will be labelled on the graph.
e.g. : helixpc graph output.csv -s -l=X9760.raw X9753 X9763
-nl | --no-legend
By default, a legend is provided on the right hand side. Since this is not very useful for standard, unicolour graphs, you may turn this off by passing –no-legend.
-nlg | --no-log
By default, the x and y coordinates will use the log_2 of the values given. This tends to give clearer graphs, as it reduces their tendency to elongate in a very thin shape, whilst still representing an accurate distribution. You can turn this off by simply passing –no-log.
-nd | --no-diagonal
By default, a diagonal x=x line will be drawn on the graph. You may turn this off by passing –no-diagonal.
input file format:
Check the example graph_input.csv The first row should specify the column titles.
The first col should contain gene_symbol


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file helixpc-1.2.1.tar.gz.
File metadata
- Download URL: helixpc-1.2.1.tar.gz
- Upload date:
- Size: 9.3 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d2c6604ac71982ce0d114da1fc5e7c5d251597cfc86e62bb1be03127e06207ad
|
|
| MD5 |
3b7d6ed2701fe82aab0f4994f9e0122e
|
|
| BLAKE2b-256 |
2318d21f660c4455f1819d845ee336737d925af8319d7d5afdf049ec89f0378b
|







