Reconstruct haplotypes and produce genealogy graphs from population data
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers


Project description
Hapsolutely





Reconstruct haplotypes and produce genealogy graphs from population data.
- Phase sequences: Reconstruct haplotypes from sequence data
- Haplotype visualization: Generate haplotype networks, genealogies and haplowebs
- Haplotype statistics: Detect fields for recombination and subset overlap
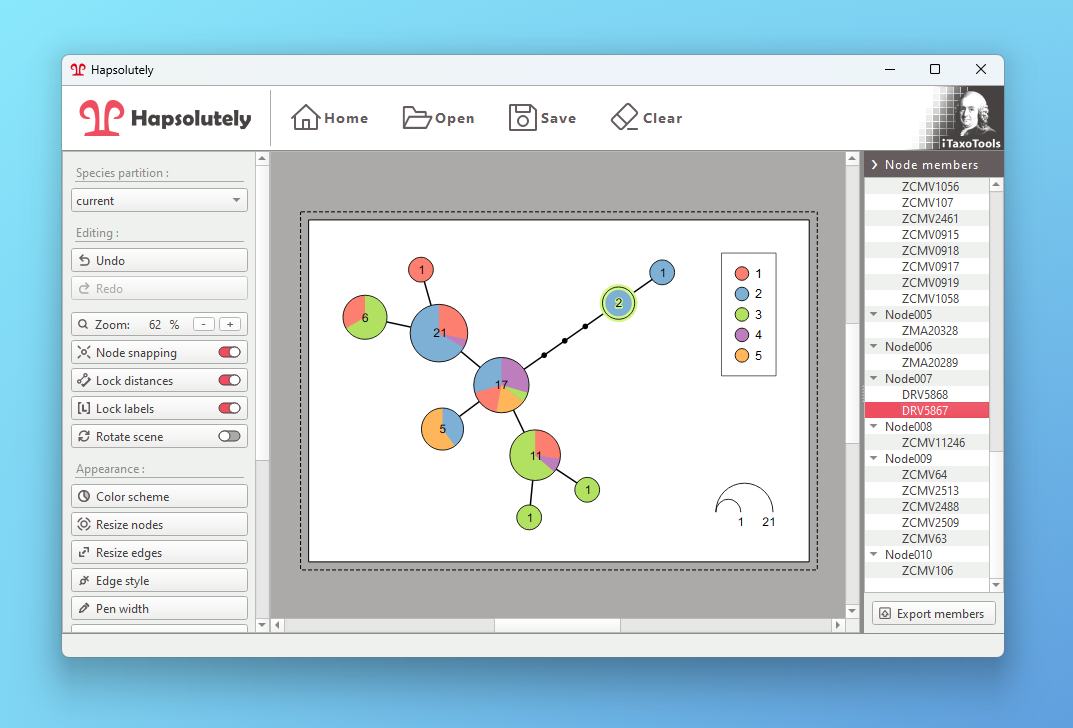
Hapsolutely is a comprehensive program that integrates ConvPhase, haplodemo, popart_networks and haplostats.
Input files can be in TSV, FASTA or SPART/XML format.
Executables
Download and run the standalone executables without installing Python.



Installation
Hapsolutely is available on PyPI. You can install it through pip:
pip install itaxotools-hapsolutely
hapsolutely
Usage
Please refer to the Hapsolutely manual for information on how to use the program.
Citations
Hapsolutely was developed in the framework of the iTaxoTools project:
Vences M. et al. (2021): iTaxoTools 0.1: Kickstarting a specimen-based software toolkit for taxonomists. - Megataxa 6: 77-92.
Sequences are phased using PHASE and SeqPHASE:
Stephens, M., Smith, N., and Donnelly, P. (2001). A new statistical method for haplotype reconstruction from population data. American Journal of Human Genetics, 68, 978--989.
Stephens, M., and Donnelly, P. (2003). A comparison of Bayesian methods for haplotype reconstruction from population genotype data. American Journal of Human Genetics, 73:1162-1169.
Flot, J.F. (2010) seqphase: a web tool for interconverting phase input/output files and fasta sequence alignments. Mol. Ecol. Resour., 10, 162–166.
Networks are generated using either of Fitchi or popart_networks:
Matschiner M (2015) Fitchi: Haplotype genealogy graphs based on the Fitch algorithm. Bioinformatics, 32:1250-252.
Leigh, JW, Bryant D (2015). PopART: Full-feature software for haplotype network construction. Methods Ecol Evol 6(9):1110-1116.
Bandelt H, Forster P, Röhl A (1999). Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16(1):37-48.
Clement M, Snell Q, Walke P, Posada D, Crandall, K (2002). TCS: estimating gene genealogies. Proc 16th Int Parallel Distrib Process Symp 2:184.
Doyle, J. J. (1995) The irrelevance of allele tree topologies for species delimitation, and a non-topological alternative. Syst. Bot., 20, 574-588.
BioPython is used to create ML/NJ trees if needed. networkx is used for laying out the initial graph.
Cock, P.J. et al., 2009. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics, 25(11), pp.1422-1423.
Hagberg, A., Swart, P. & S Chult, D., 2008. Exploring network structure, dynamics, and function using NetworkX.
Project details
Verified details
These details have been verified by PyPIProject links
GitHub Statistics
Maintainers


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file itaxotools_hapsolutely-0.2.3.tar.gz.
File metadata
- Download URL: itaxotools_hapsolutely-0.2.3.tar.gz
- Upload date:
- Size: 506.4 kB
- Tags: Source
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.12.9
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
c7d6bb013102bd4ffea86421b8759727ebbb082545fcd5832ff5bb19da8e9bc5
|
|
| MD5 |
99e77a4e46327a68486cc933455e4ff4
|
|
| BLAKE2b-256 |
baf6e00e99f4d095b7b8d145b4d4f20cf778a83bc4b31e1c5a0d86334cf69d81
|
Provenance
The following attestation bundles were made for itaxotools_hapsolutely-0.2.3.tar.gz:
Publisher:
deploy.yml on iTaxoTools/Hapsolutely
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
itaxotools_hapsolutely-0.2.3.tar.gz -
Subject digest:
c7d6bb013102bd4ffea86421b8759727ebbb082545fcd5832ff5bb19da8e9bc5 - Sigstore transparency entry: 230379018
- Sigstore integration time:
-
Permalink:
iTaxoTools/Hapsolutely@ed49ef00f0e21a2b1c60341db7d7383c1ab2d54b -
Branch / Tag:
refs/tags/v0.2.3 - Owner: https://github.com/iTaxoTools
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
deploy.yml@ed49ef00f0e21a2b1c60341db7d7383c1ab2d54b -
Trigger Event:
push
-
Statement type:
File details
Details for the file itaxotools_hapsolutely-0.2.3-py3-none-any.whl.
File metadata
- Download URL: itaxotools_hapsolutely-0.2.3-py3-none-any.whl
- Upload date:
- Size: 107.4 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.12.9
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
834961386c805ddf6a474be3bb5817ab1de1184e0191ee15aee0f5b3a0d781e3
|
|
| MD5 |
2d960ba85212aab86e650855f5c0960d
|
|
| BLAKE2b-256 |
496e6b0a012e1ad35bd6434b521ed6d13918128fab8da7de698bb9a6d21ad564
|
Provenance
The following attestation bundles were made for itaxotools_hapsolutely-0.2.3-py3-none-any.whl:
Publisher:
deploy.yml on iTaxoTools/Hapsolutely
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
itaxotools_hapsolutely-0.2.3-py3-none-any.whl -
Subject digest:
834961386c805ddf6a474be3bb5817ab1de1184e0191ee15aee0f5b3a0d781e3 - Sigstore transparency entry: 230379023
- Sigstore integration time:
-
Permalink:
iTaxoTools/Hapsolutely@ed49ef00f0e21a2b1c60341db7d7383c1ab2d54b -
Branch / Tag:
refs/tags/v0.2.3 - Owner: https://github.com/iTaxoTools
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
deploy.yml@ed49ef00f0e21a2b1c60341db7d7383c1ab2d54b -
Trigger Event:
push
-
Statement type:







