A software for RNA-seq investigation using k-mer decomposition


Project description



Introduction:
This tool was developed to identify and quantify the occurence of single nucleotide variants, insertions, deletions and duplications in RNA-seq data. Contrary to most tools that try to report all variants in a complete genome, here we instead propose to focus the analysis on small regions of interest.
Given a reference sequence (typically a few hundred base pairs) around a known or suspected mutation in a gene of interest, all possible sequences that can be be created between the two end k-mers according to the sequenced reads will be reported. A ratio of variant allele vs WT will be computed for each possible sequence constructed.
Citing:
Targeted variant detection using unaligned RNA-Seq reads. Life science Alliance 2019 Aug 19;2(4); doi: https://doi.org/10.26508/lsa.201900336
Target variant detection in leukemia using unaligned RNA-Seq reads. bioRxiv 295808; doi: https://doi.org/10.1101/295808
Install:
Using pip (recommended)
python3 -m venv $HOME/.virtualenvs/km
source $HOME/.virtualenvs/km/bin/activate
pip install --upgrade pip setuptools wheel
pip install km-walkRequirements:
Python 3.8.0 or later with pip installed.
Usage:
The virtual environment needs to be loaded each time you open a new terminal, with this command:
$ source $HOME/.virtualenvs/km/bin/activateTest:
4bp insertion in NPM1
$ cd [your_km_folder]
$ km find_mutation ./data/catalog/GRCh38/NPM1_4ins_exons_10-11utr.fa ./data/jf/02H025_NPM1.jf | km find_report -t ./data/catalog/GRCh38/NPM1_4ins_exons_10-11utr.fa
Sample Region Location Type Removed Added Abnormal Normal Ratio Min_coverage Exclu_min_cov Variant Target InfoVariant_sequence Reference_sequence
./data/jf/02H025_NPM1.jf chr5:171410540-171410543 chr5:171410544 ITD 0 4 | 4 2870.6 3055.2 0.484 2428 /TCTG NPM1_4ins_exons_10-11utr vs_ref AATTGCTTCCGGATGACTGACCAAGAGGCTATTCAAGATCTCTGTCTGGCAGTGGAGGAAGTCTCTTTAAGAAAATAGTTTAAA AATTGCTTCCGGATGACTGACCAAGAGGCTATTCAAGATCTCTGGCAGTGGAGGAAGTCTCTTTAAGAAAATAGTTTAAA
./data/jf/02H025_NPM1.jf - Reference 0 0 0.0 2379.0 1.000 2379 - NPM1_4ins_exons_10-11utr vs_ref
# To display kmer coverage
$ km find_mutation ./data/catalog/GRCh38/NPM1_4ins_exons_10-11utr.fa ./data/jf/02H025_NPM1.jf -gITD of 75 bp
$ cd [your_km_folder]
$ km find_mutation ./data/catalog/GRCh38/FLT3-ITD_exons_13-15.fa ./data/jf/03H116_ITD.jf | km find_report -t ./data/catalog/GRCh38/FLT3-ITD_exons_13-15.fa
Sample Region Location Type Removed Added Abnormal Normal Ratio Min_coverage Exclu_min_cov Variant Target Info Variant_sequence Reference_sequence
./data/jf/03H116_ITD.jf - Reference 0 0 0.0 443.0 1.000 912 - FLT3-ITD_exons_13-15 vs_ref
./data/jf/03H116_ITD.jf chr13:28034105-28034179 chr13:28034180 ITD 0 75 | 75 417.6 1096.7 0.276 443 /AACTCCCATTTGAGATCATATTCATATTCTCTGAAATCAACGTAGAAGTACTCATTATCTGAGGAGCCGGTCACC FLT3-ITD_exons_13-15 vs_ref CTTTCAGCATTTTGACGGCAACCTGGATTGAGACTCCTGTTTTGCTAATTCCATAAGCTGTTGCGTTCATCACTTTTCCAAAAGCACCTGATCCTAGTACCTTCCCAAACTCTAAATTTTCTCTTGGAAACTCCCATTTGAGATCATATTCATATTCTCTGAAATCAACGTAGAAGTACTCATTATCTGAGGAGCCGGTCACCAACTCCCATTTGAGATCATATTCATATTCTCTGAAATCAACGTAGAAGTACTCATTATCTGAGGAGCCGGTCACCTGTACCATCTGTAGCTGGCTTTCATACCTAAATTGCTTTTTGTACTTGTGACAAATTAGCAGGGTTAAAACGACAATGAAGAGGAGACAAACACCAATTGTTGCATAGAATGAGATGTTGTCTTGGATGAAAGGGAAGGGGC CTTTCAGCATTTTGACGGCAACCTGGATTGAGACTCCTGTTTTGCTAATTCCATAAGCTGTTGCGTTCATCACTTTTCCAAAAGCACCTGATCCTAGTACCTTCCCAAACTCTAAATTTTCTCTTGGAAACTCCCATTTGAGATCATATTCATATTCTCTGAAATCAACGTAGAAGTACTCATTATCTGAGGAGCCGGTCACCTGTACCATCTGTAGCTGGCTTTCATACCTAAATTGCTTTTTGTACTTGTGACAAATTAGCAGGGTTAAAACGACAATGAAGAGGAGACAAACACCAATTGTTGCATAGAATGAGATGTTGTCTTGGATGAAAGGGAAGGGGCBootstrap:
Or you can run easy_install.sh which installs km in a virtual environement and test it as shown above. Running the script as is will install km in a virtual environment in: $HOME/.virtualenvs/km.
./easy_install.shFrom source code
km can be executed directly from source code.
Requirements:
Python 3.6.0 or later
Jellyfish 2.2 or later with Python bindings (or pyJellyfish module).
Usage:
$ cd [your_km_folder]
$ python -m km find_mutation ./data/catalog/GRCh38/NPM1_4ins_exons_10-11utr.fa ./data/jf/02H025_NPM1.jf | km find_report -t ./data/catalog/GRCh38/NPM1_4ins_exons_10-11utr.faDesign your target sequence:
km is designed to make targeted analysis based on target sequences. These target sequences need to be designed and given to km as input.
A target sequence is a nucleotide sequence saved in a fasta file. Some target sequences are provided in catalog.
To fit your specific needs, you will have to create your own target sequences.
On generic cases, you can follow some good practices described below:
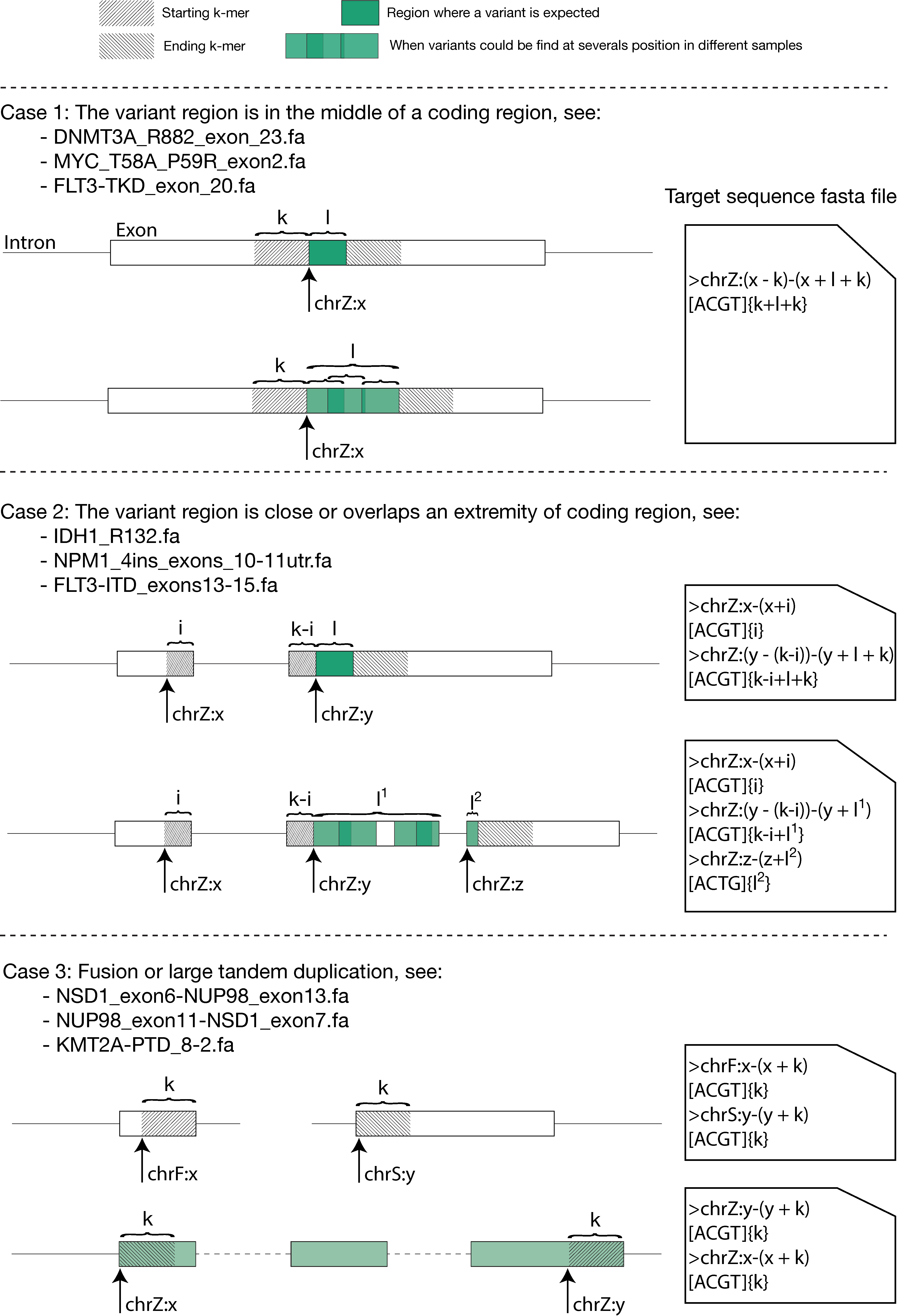
A web portal is available to assist you in the creation of your target sequences (for cases 1 and 2).
km-target: https://bioinfo.iric.ca/km-target/
You could also extract nucleotide sequences from genome using severals methods, two of them are discribe below:
Display help:
$ km -h
usage: PROG [-h] {find_mutation,find_report,linear_kmin,min_cov} ...
positional arguments:
{find_mutation,find_report,linear_kmin,min_cov}
sub-command help
find_mutation Identify and quantify mutations from a target sequence
and a k-mer database.
find_report Parse find_mutation output to reformat it in tabulated
file more user friendly.
linear_kmin Find min k length to decompose a target sequence in a
linear graph.
min_cov Compute coverage of target sequences.
optional arguments:
-h, --help show this help message and exitkm’s tools overview:
For more detailed documentation click here.
find_mutation:
This is the main tool of km, to identify and quantify mutations from a target sequence and a k-mer jellyfish database.
$ km find_mutation -h
$ km find_mutation [your_fasta_targetSeq] [your_jellyfish_count_table]
$ km find_mutation [your_catalog_directory] [your_jellyfish_count_table]find_report:
This tool parse find_mutation output to reformat it in more user friendly tabulated file.
$ km find_report -h
$ km find_report -t [your_fasta_targetSeq] [find_mutation_output]
$ km find_mutation [your_fasta_targetSeq] [your_jellyfish_count_table] | km find_report -t [your_fasta_targetSeq]min_cov:
This tools display some k-mer’s coverage stats of a target sequence and a list of jellyfish database.
$ km min_cov -h
$ km min_cov [your_fasta_targetSeq] [[your_jellyfish_count_table]...]linear_kmin:
Length of k-mers is a central parameter:
To produce a linear directed graph from the target sequence.
To avoid false-positive. find_mutation shouldn’t be use on jellyfish count table build with k<21 bp (we recommand k=31 bp, by default)
linear_kmin tool is design to give you the minimun k length to allow a decomposition of a target sequence in a linear graph.
$ km linear_kmin -h
$ km linear_kmin [your_catalog_directory]Runing km on a real sample from downloaded fastq:
In the example folder you can find a script to help you to run a km analysis on one Leucegene sample.


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file km_walk-2.2.2.tar.gz.
File metadata
- Download URL: km_walk-2.2.2.tar.gz
- Upload date:
- Size: 32.0 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/5.1.0 CPython/3.12.2
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
8fe2f4507a6f2a4491201a7aa5c786ca3bc50c9d6ac8ebbdc691592b7c44e80e
|
|
| MD5 |
30b3b03f2353125d3077ef7a0dcee464
|
|
| BLAKE2b-256 |
9107224393dde359158ed0a29081347b3c8e0550bcc046d5fa20d726ca31923c
|
File details
Details for the file km_walk-2.2.2-py3-none-any.whl.
File metadata
- Download URL: km_walk-2.2.2-py3-none-any.whl
- Upload date:
- Size: 33.5 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/5.1.0 CPython/3.12.2
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
2d80999f3ffd5cd17fd330ca166cc2531ea09f6e08f5be06970765cb028d4cdd
|
|
| MD5 |
3fbbf19991bff0c340b7734a0d48fc4a
|
|
| BLAKE2b-256 |
f6797e9ccf345abfe10c2f8daed772279c3fc089266c97d0078c160d427cc93d
|







