tools for analyzing and exploring genetic relationships

Project description









lineage
tools for analyzing and exploring genetic relationships 🧬
lineage strives to be an easy-to-use and accessible open-source library for genetic genealogy
Features
Shared DNA Analysis
- Find shared DNA between individuals using genetic maps from the HapMap Project and 1000 Genomes Project
- Compute centiMorgans (cMs) of shared DNA with configurable thresholds
- Detect IBD1 (half-identical) and IBD2 (fully identical) regions
- Visualize shared DNA segments across all chromosomes
- Find discordant SNPs inconsistent with Mendelian inheritance patterns
Shared Gene Analysis
- Identify genes shared between individuals with the same genetic variations
- Determine which genes produce the same proteins across related individuals
Synthetic Data Generation
- Generate realistic synthetic genotype data for related individuals
- Create parent-child pairs with proper single-allele inheritance
- Create sibling pairs with realistic meiotic recombination patterns
Supported Genotype Files
lineage supports all genotype files supported by snps.
Installation
lineage is available on the
Python Package Index. Install lineage (and its required
Python dependencies) via pip:
$ pip install lineage
Examples
The examples below demonstrate the core features of lineage. For detailed explanations of
genetic concepts like IBD (Identity By Descent), centiMorgans, and how to interpret results,
see the Concepts Guide.
Optional: To see file save notifications, configure logging before running examples:
import logging
logging.basicConfig(level=logging.INFO, format='%(message)s')
To try these examples, first generate some sample data:
>>> from lineage import Lineage
>>> l = Lineage()
>>> paths = l.create_example_datasets()
Load Individuals
Load genotype files and create Individual objects:
>>> parent = l.create_individual('Parent', paths['parent'])
>>> child = l.create_individual('Child', paths['child'])
Each Individual inherits from snps.SNPs,
providing access to all SNPs properties and methods:
>>> parent.build
37
>>> parent.assembly
'GRCh37'
>>> parent.count
899992
Find Discordant SNPs
Identify SNPs inconsistent with Mendelian inheritance between parent and child:
>>> discordant_snps = l.find_discordant_snps(parent, child, save_output=True) # doctest: +SKIP
The example datasets include a small number of simulated genotyping errors (~0.01%) to demonstrate discordant SNP detection.
Find Shared DNA
Parent-Child Example
Compute shared DNA segments and generate a visualization:
>>> results = l.find_shared_dna([parent, child]) # doctest: +SKIP
For parent-child relationships, all shared DNA appears on one chromosome only (IBD1), representing the chromosome inherited from that parent (~3400-3700 cM total):
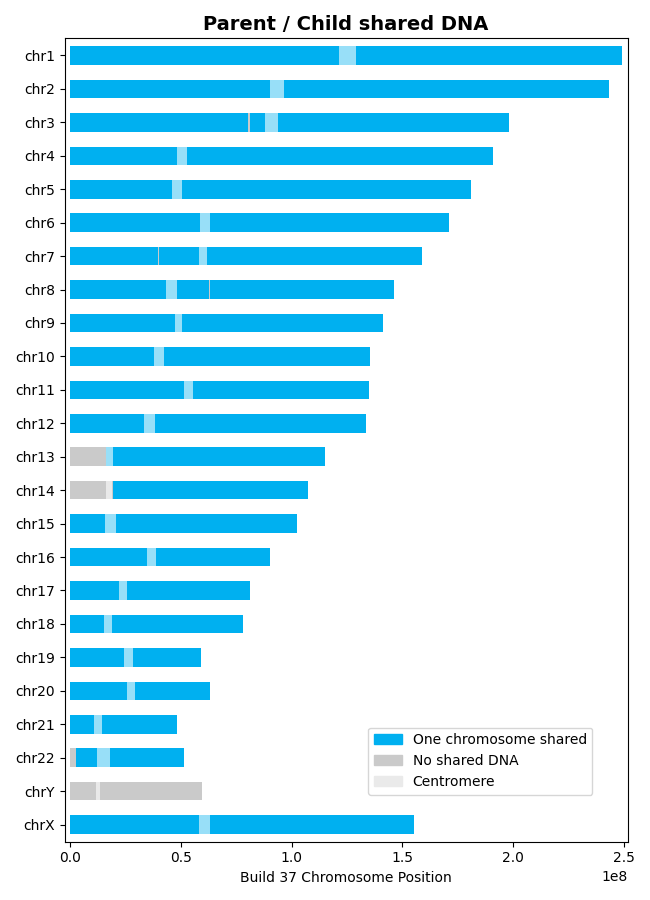
Sibling Example
Analyze siblings using a population-specific genetic map:
>>> sibling1 = l.create_individual('Sibling1', paths['sibling1']) # doctest: +SKIP
>>> sibling2 = l.create_individual('Sibling2', paths['sibling2']) # doctest: +SKIP
>>> results = l.find_shared_dna([sibling1, sibling2], shared_genes=True, genetic_map="CEU") # doctest: +SKIP
Siblings share DNA on one chromosome (IBD1) and both chromosomes (IBD2), reflecting segments where they inherited the same DNA from one or both parents:
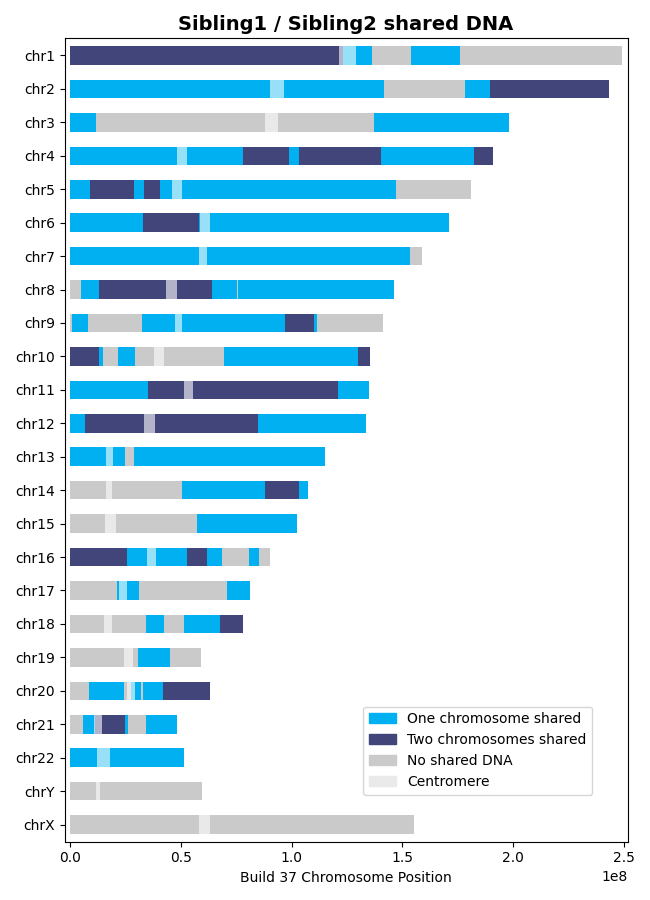
Documentation
Documentation is available here.
Acknowledgements
Thanks to Whit Athey, Ryan Dale, Binh Bui, Jeff Gill, Gopal Vashishtha, CS50. This project was historically validated using data from openSNP.
lineage incorporates code and concepts generated with the assistance of various
generative AI tools (including but not limited to ChatGPT,
Grok, and Claude). ✨
License
lineage is licensed under the MIT License.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file lineage-4.5.0.tar.gz.
File metadata
- Download URL: lineage-4.5.0.tar.gz
- Upload date:
- Size: 71.4 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.14.2
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
5f756d3b4057600604c41847bd918b46f4180d54df6cae674345cf335bd39f13
|
|
| MD5 |
b3e78e5eb27818d9bebf7b583e5b7a74
|
|
| BLAKE2b-256 |
f13ba444acf043639b595c112f5af8e520a5457d5df53f4c397a7490aa4f7d70
|
File details
Details for the file lineage-4.5.0-py3-none-any.whl.
File metadata
- Download URL: lineage-4.5.0-py3-none-any.whl
- Upload date:
- Size: 27.5 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.14.2
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
60c2e1c3b50771c54f18620ab76882dab0b1ae36ef8edd57f60aab5392f090a6
|
|
| MD5 |
bb3f52245e0ae94038c273fb1ca526d1
|
|
| BLAKE2b-256 |
9dbab97d3c59af1ff9815a4dc27227e57d86b47a59d7b08f4e2a8c5be32079c9
|







