A small Python package to manipulate complex lipids.

Project description
liputils
A small Python package to manipulate complex lipids.
Overview
liputils makes it easy to strip fatty acids-like residues from individual molecular lipids. This is done by liputils by parsing the lipid string identifier.
Check it out! We have a paper out in Scientific Reports, with detailed step-by-step installation and usage protocols, and real use cases implemented and discussed:
liputils: a Python module to manage individual fatty acid moieties from complex lipids
Stefano Manzini, Marco Busnelli, Alice Colombo, Mostafa Kiamehr, Giulia Chiesa
PMID: 32770020 PMCID: PMC7415148 DOI: 10.1038/s41598-020-70259-9


Tracking individual residues is is particularly useful when wanting to track how the carbon chains move across the lipidome, independently from where they are attached to. For instance, it is possible to see if the general trend of long carbon residues in the plasma matches the data available from the dietary treatment.
The swiftest way to convert your lipidomic data into a residue count (usually given in submolar fractions) is by using liputils' built-in GUI:
>>> from liputils import GUI
>>> GUI()
This brings up the GUI (here's what it looks like in MacOS):
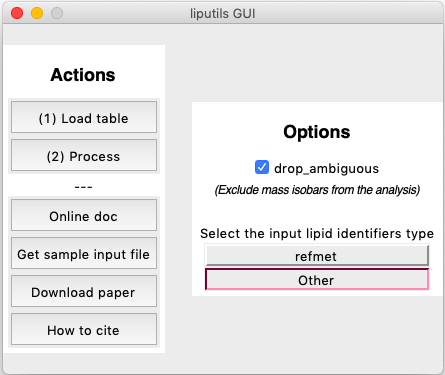
The GUI acts as a wrapper for make_residues_table(), enabling a fast two-click conversion of any source table. All is required is to 1) choose the table to convert and 2) Process the table. When hitting (2) Process, the user is first asked to choose where to put and how to call the newly produced table, then everything happens automatically. That's it! Try running it with default settings (the ones the GUI starts up with) on the sample data.
Of course we're taking a lot for granted, but this was just a quick start, dig down in the doc to find out things like how data needs to be shaped or what lipid identifiers are supported, as well as what else liputils can do for you within a Python REPL.
The Lipid class takes care of extracting information from the lipid name:
>>> from liputils import Lipid
>>> l = Lipid("PG 18:1/20:1", amount=0.012512)
>>> l.lipid_class()
'PG'
>>> l.name
'PG 18:1/20:1'
>>> l.residues()
(['18:1', '20:1'], 1)
>>> l.molecules
7534902640.2784
>>> l.amount # the original value is stored here
0.012512
The number of molecules is calculated from the amount parameter, defaulting to picomoles. This can be changed:
>>> l = Lipid("PG 18:1/20:1", amount=0.012512, unit="femtomoles")
>>> l.molecules
7534902.640278401
In the case of unresolved ambiguities of the lipid isomers, it is possible to either extract all of them and choose how to manage that information by taking into consideration how many ambiguities there are:
>>> l = Lipid("TAG 48:2 total (14:0/16:0/18:2)(14:0/16:1/18:1)(16:0/16:1/16:1)")
>>> l.residues()
(['14:0', '16:0', '18:2', '14:0', '16:1', '18:1', '16:0', '16:1', '16:1'], 3)
Or, it is possible to reject non unambiguous lipids altogether by calling .residues() with the drop_ambiguous parameter:
>>> l = Lipid("PG 18:1/20:1")
>>> l.residues(drop_ambiguous=True)
(['18:1', '20:1'], 1)
>>> l = Lipid("TAG 48:2 total (14:0/16:0/18:2)(14:0/16:1/18:1)(16:0/16:1/16:1)")
>>> l.residues(drop_ambiguous=True)
([], 0)
Data formats
liputils accepts a generic lipid format in the form of CLASS N:N/M:M/../.. or CLASS(N:N/M:M/../..)(other mass isomers), or fully RefMet-compliant residue naming. If unsure about your data format, you can try and batch-translate your lipid IDs with RefMet's online translator. By adhering to RefMet's nomenclature, any compliant lipid name will be properly managed by liputils's method .refmet_residues():
>>> lip1 = Lipid("octadecatrienoic acid")
>>> lip2 = Lipid("linolenic acid")
>>> lip3 = Lipid("FA(18:3)")
>>> lip1.refmet_residues()
(['18:3'], 1)
>>> lip2.refmet_residues()
(['18:3'], 1)
>>> lip3.refmet_residues()
(['18:3'], 1)
Composite compounds can also be fed to liputils:
>>> l = Lipid("linoleyl palmitate")
>>> l.refmet_residues()
(['18:1', '16:0'], 1)
One-step lipidomics data conversion
Lipidomics data should be loaded in a pandas.DataFrame table. The accepted format is a vertical index with lipid names, and samples in column. Just like this:
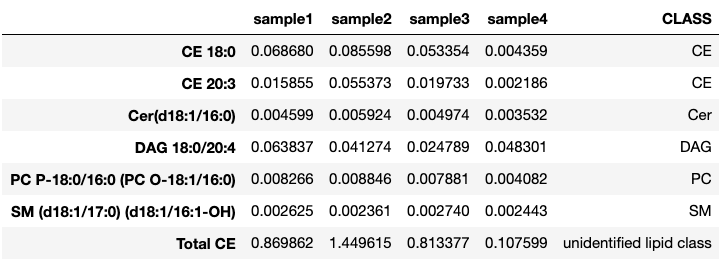
make_residues_table() will take care of dropping non-numerical columns, as well as to trim the lipid list of elements that should not be processed, like total lipid class counts. These can be further specified through the unwanted parameter.
Getting the transformed table is super easy:
from liputils import make_residues_table
# df is out dataframe
res = make_residues_table(df)
In res, we will find the resulting table:
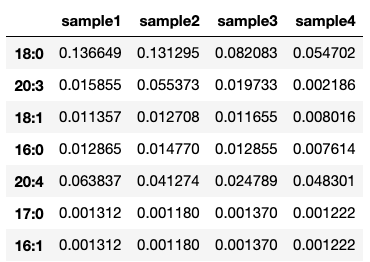
To focus on particular residues, it is possible to mix saturated() and max_carbon() to dictate which residues to keep in the index and which to discard:
from liputils import saturated, max_carbon
>>> saturated("12:0")
True
>>> max_carbon("12:0", 16)
True
>>> max_carbon("21:3", 16)
False
>>> my_lipids = ["12:0", "17:1", "24:0", "24:1", "24:2", "26:3"]
>>> [not saturated(x) and max_carbon(x, 24) for x in my_lipids]
[False, True, False, True, True, False]
For further info, don't forget to investigate around:
help(make_residues_table)
Parameters
==========
dataframe: a pandas dataframe of data. Lipid names as index, and samples as columns
(just unlike sklearn wants it, but as you might get it from Tableau software
tables. Just dataframe.T your table - that would just do the trick).
drop_ambiguous: <bool> don't take isobars into consideration. Defaults to False. If True,
each residue is divided by its uncertainty.
name: <str> a tag that gets attached to the returned dataframe, so you can use it
to save it afterwards. The tag is found in the .name attribute.
replace_nan: <object> the object you would like to replace your missing values with.
It can be set to False, but I would suggest against that.
cleanup: <bool> Whether to perform a cleanup of unwanted lipids that can be present
in the index. Unwanted strings are read from the 'unwanted' parameter. Defaults
to True
absolute_amount <bool> Wheter to count the individual number of residues, rather to
sticking to the same units found in the original table. Defaults to False
unwanted: <list> <set> <tuple> Strings that must be removed from the lipid index. Defaults
to ["total", "fc", "tc"]
returns:
========
pandas DataFrame


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file liputils-0.16.2.tar.gz.
File metadata
- Download URL: liputils-0.16.2.tar.gz
- Upload date:
- Size: 19.2 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.4.0 importlib_metadata/3.7.3 packaging/20.8 pkginfo/1.7.0 requests/2.25.1 requests-toolbelt/0.9.1 tqdm/4.56.0 CPython/3.8.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
aac1689431686cdd9a6c42dbc114c9cfd4b540f884dfd4971b37fa6fe7623fa8
|
|
| MD5 |
0a96bbd86b42162e66c3217f00cc03f8
|
|
| BLAKE2b-256 |
df33f9f8aa45c79204dde4beed64fcdfdcbbe3ae3c751bc77258c0fdffa533c4
|







