MESSI: Multi Ensamble Strategy for Structural Elucidation

Project description
MESSI
Multi Ensamble Strategy for Structural identification
Authors: Ariel M. Sarotti & María M. Zanardi
This repository contains all codes and data required to run MESSI calculations.
Description
MESSI is a Python program to compute Multi Ensamble DP4+ probabilities in the stereochemical assignament of new organic compounds.
Installation Requirements
MESSI.py needs python 3.8 or later to work. The module can be installed by console using:
pip3 install messi_nmr
Usage by console: messi
Alternatively can generate a shortcut on the desktop messi.py, which allows direct execution of the program without the use of a console, using: messi_exe
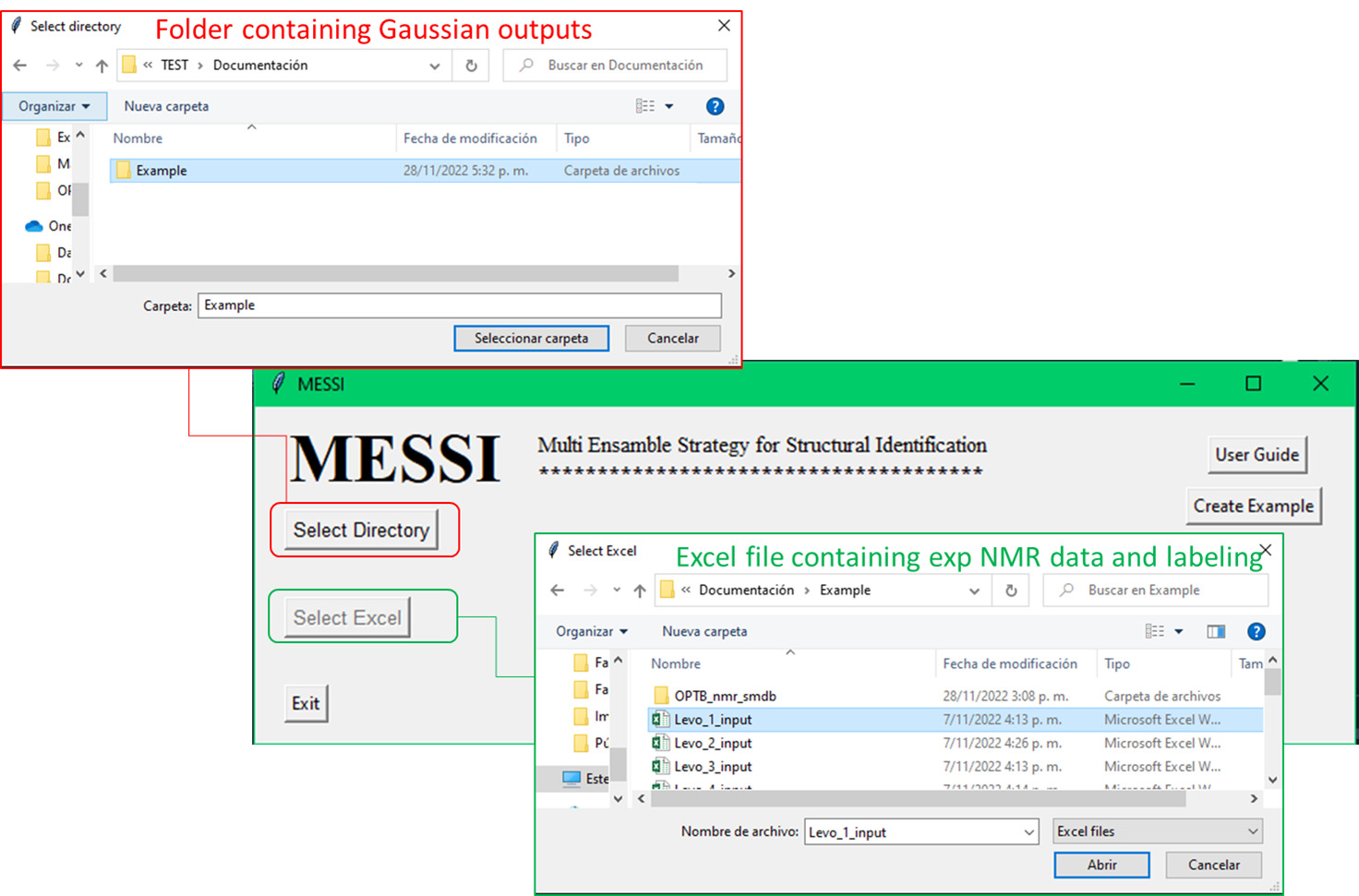
In order to test the correct software operation is recommended to run the provided example, which could be download by clicking the buttom
Create Example. This will create a folder nameExample_messi_nmrin desktop containing all the files needed by de use of MESSI.
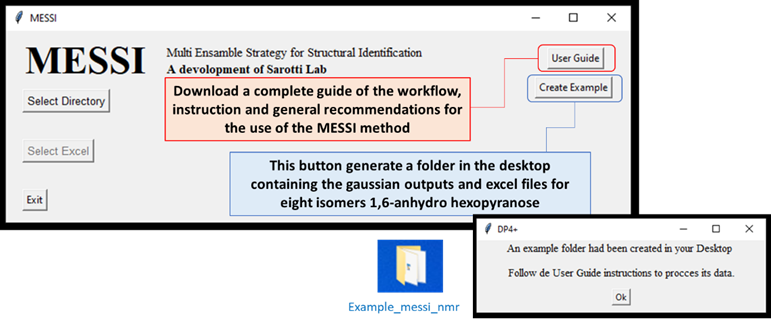
User Guide
Terms of use. You need to create a folder containing the following files:
1) The outputs of the NMR and SCF calculations (all conformers for all isomers).
2) An excel file containing the experimental data and the labels of each nucleus associated with each experimental value.
1) The output files: must be named following the next convention: n_*_m*.log or .out, where n identifies the ith isomer, ranging from 1 to N where N is the number of candidate structures under study, and m indicate the conformer number. For instance:
1_NewNatProd_c01.log (Conformer 1 of isomer 1 of a compound named NewNatProd)
1_NewNatProd_c02.log (Conformer 2 of isomer 1 of a compound named NewNatProd)
2_NewNatProd_c01.log (Conformer 1 of isomer 2 of a compound named NewNatProd)
2_NewNatProd_c02.log (Conformer 2 of isomer 2 of a compound named NewNatProd)
The NMR and SCRF/SMD energies calculation could be in the same or different outputs. If they are separated, both file must begin with the same name (
n_*_m*) and a suffix must be added in order to differentiate the calculation type. If the number of files for NMR calculations does not match the number of SMD energy calculations the script will not run.
The script handles outputs from Gaussian 09 and 16.
Note: Name files adequately is essential in order to match SMD energy with the NMR data for each conformer.
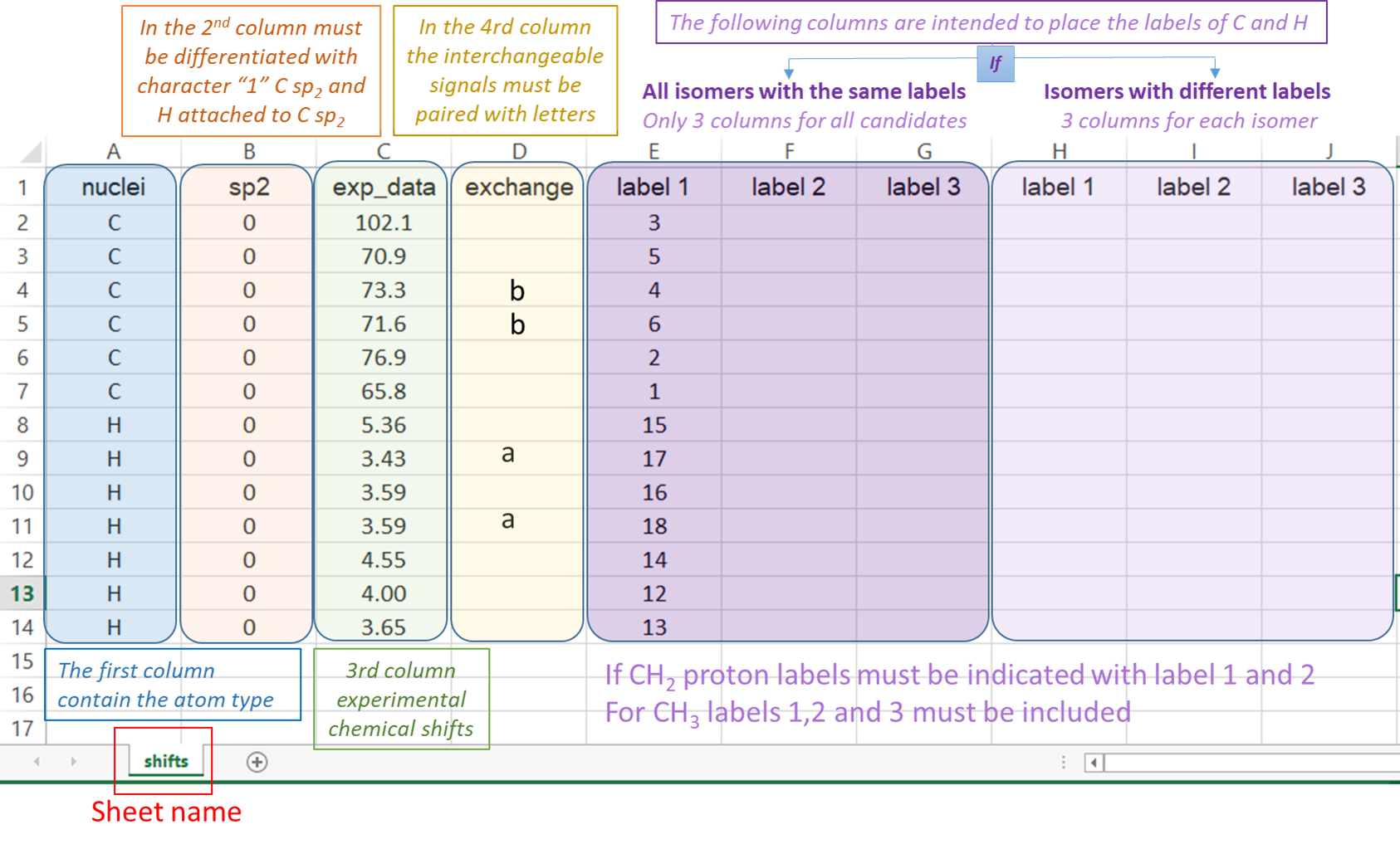
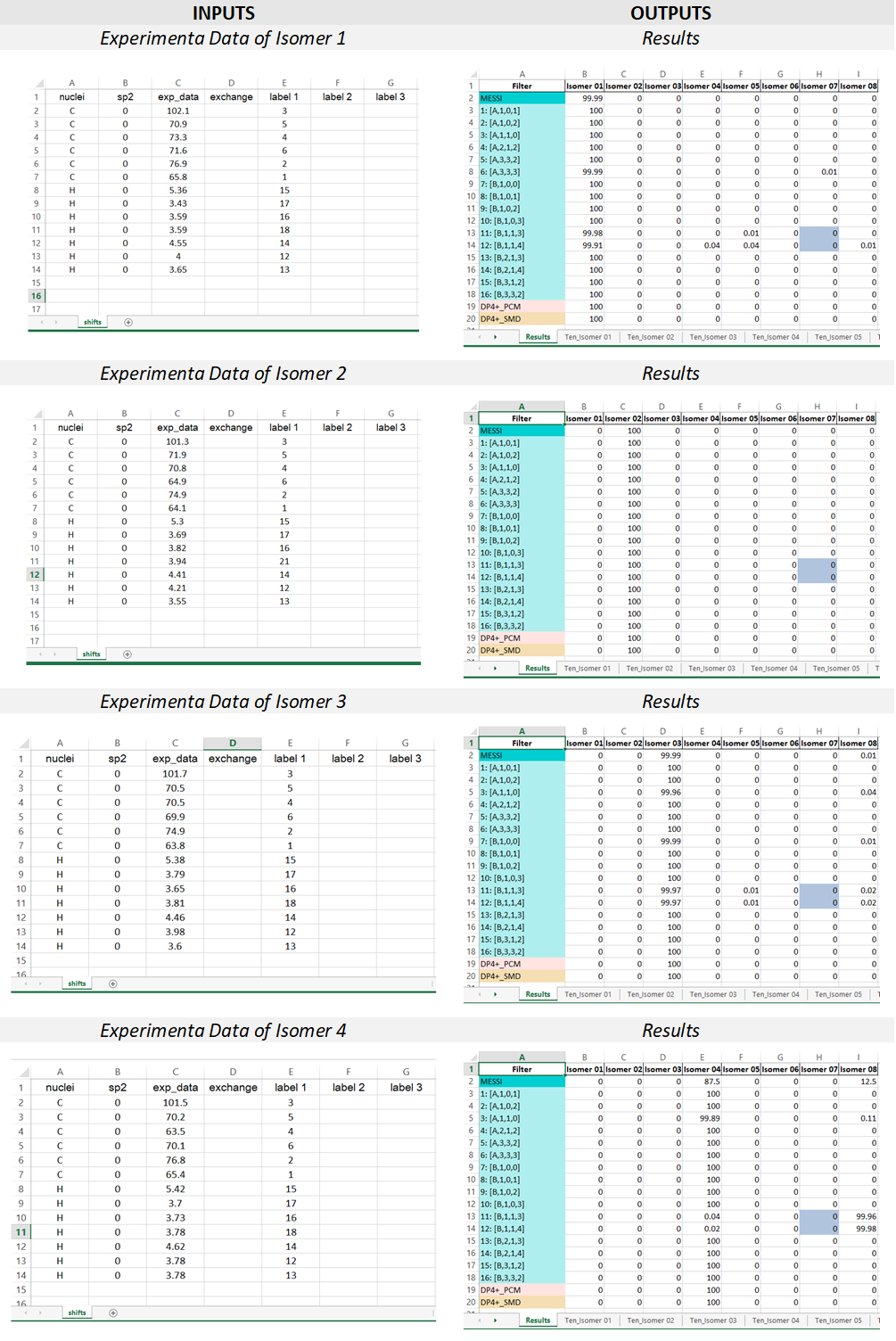
2) The input Excel file: The experimental data and the labels of the candidate structures must be placed in an excel file following the next rules. The excel file should be constituted by one sheet; containing the data of the NMR chemical shifts (named ‘shifts’).
“shifts” sheet Structure: the first column “nuclei” contain the identity of the atom ‘c or C’ for 13C and ‘h or H’ for hydrogen atoms. The second column “sp2” serves to indicate 0 (for sp3 C or H attached to) or 1 (for sp2 and sp). The third column “exp_data” contains the experimental chemical shifts. The column “exchange” allows to indicate interchangeable signals (for example, two diastereotopic hydrogens). Any character can be used to indicate a pair of interchangeable signals, which will cause that the experimental and calculated values to be ordered upside-down. When dealing with more than one pair of interchangeable signals, different characters should be used to differentiate them. For example, it can be used the letter “a to indicate one pair, and the letter “b” to indicate the other pair. The following columns are intended to place the labels of the nuclei associated to the corresponding chemical shift. If two or more values are added in that region, the isotropic shielding values will be averaged (as in the case of methyl groups or equivalent methylene groups). If the isomers under study have different labeling schemes (as in the case of constitutional isomers), three columns for each isomer should be provided as indicated below.
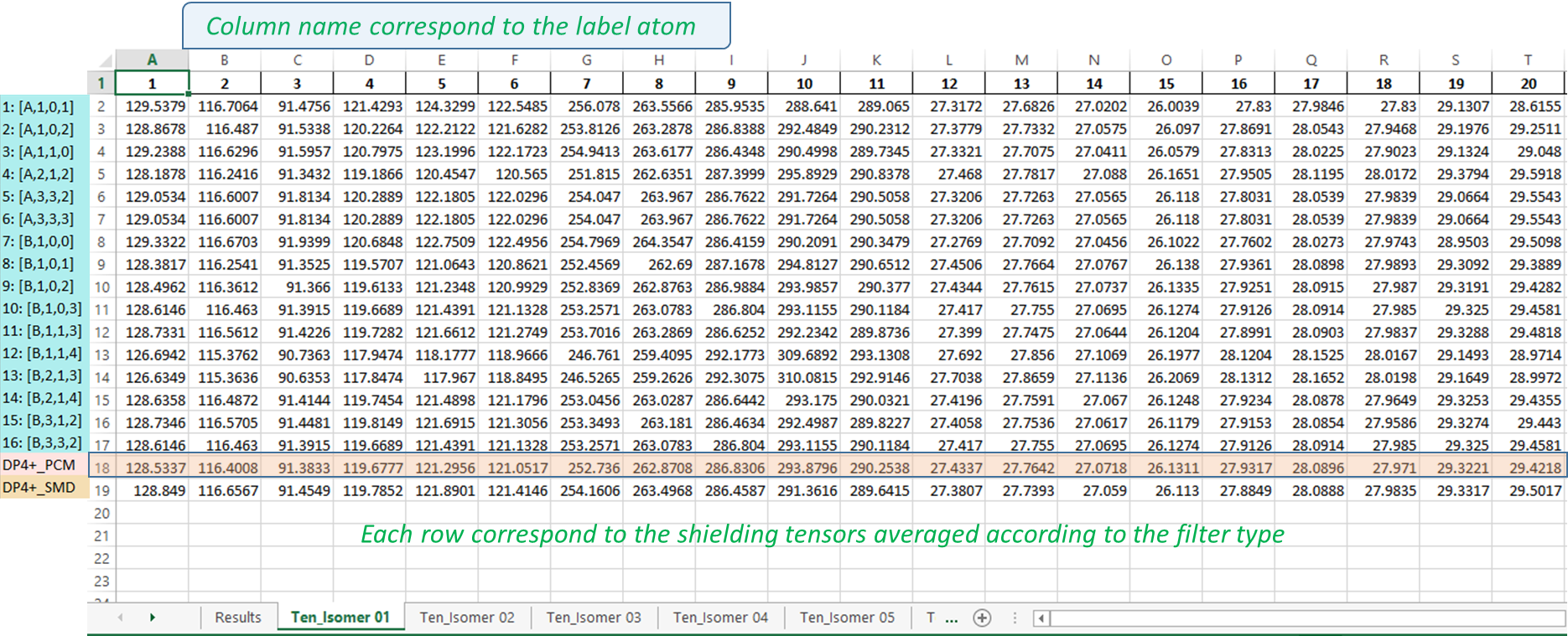
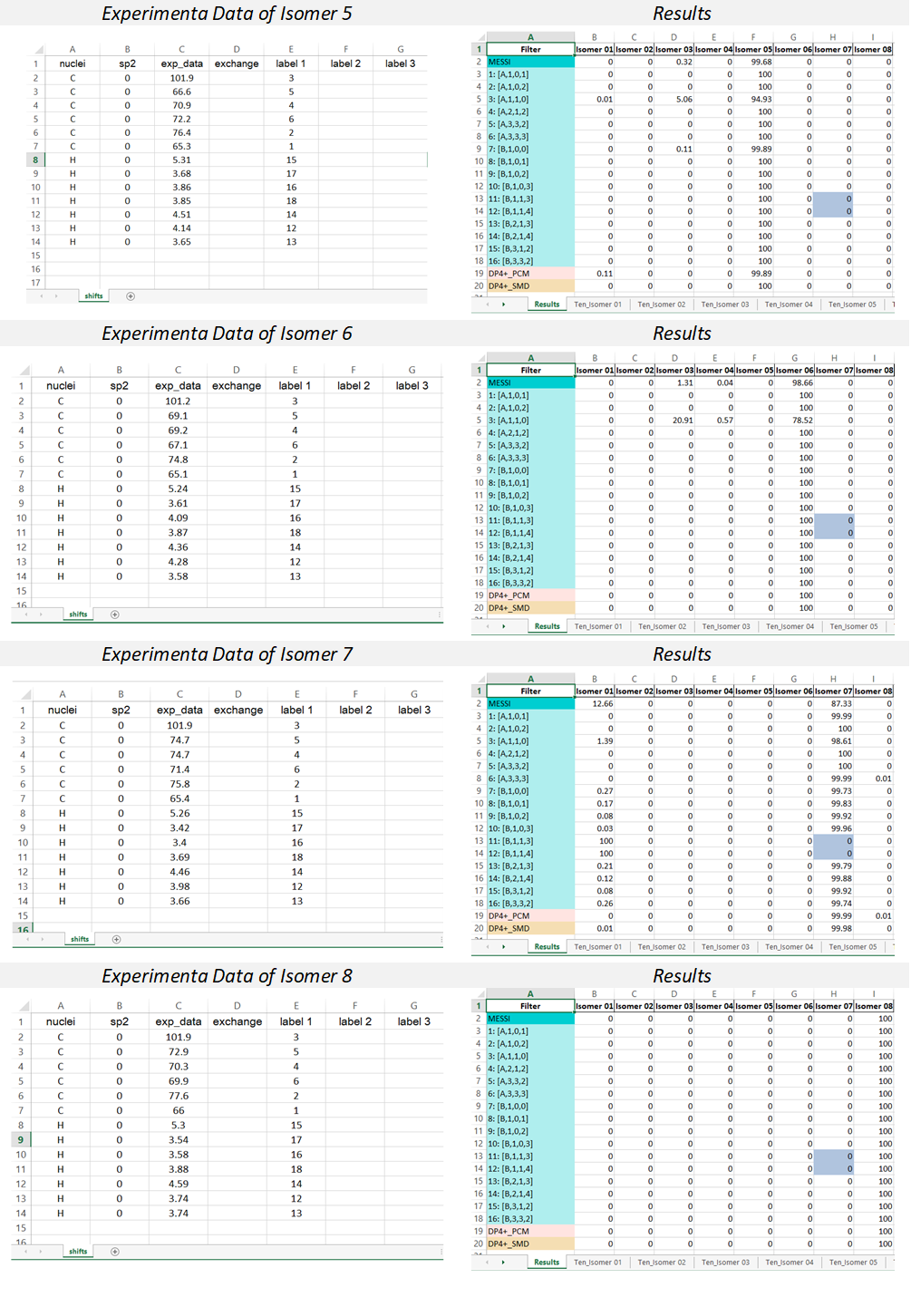
3) The output excel file: once the messi.py is executed, a file named ‘MESSI_Results.xlsx’ is created in the folder containing the Gaussian outputs. The file contains n+1 sheets where n is the number candidates structures:
Results sheet: contain the PCM-DP4+ (row 19, standard DP4+), SMD-DP4+ (row 20, standard DP4+ using the energies computed at the SMD/B3LYP/6-31+G** level), and the DP4+ results computed for the selected 16 ensembles (rows 3-18). The averaged values of those 16 calculations (MESSI.) are shown in row 2. If both 1H and 13C are used, the probabilities shown correspond to the full DP4+ results. In case only 1H, or 13C, data are used (not recommended), the probabilities shown correspond to H-DP4+ or C-DP4+ values, respectively.
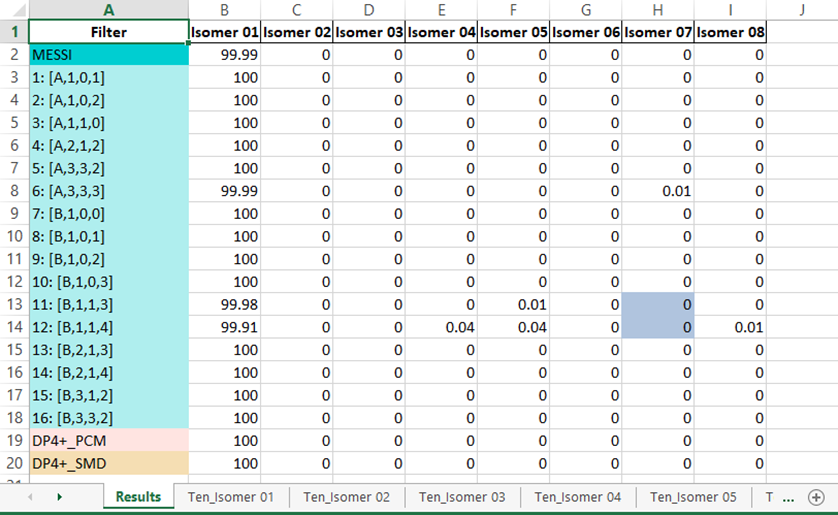
NOTE: It is important to point out that ensembles 4, 11 y 12 are created by removing all conformations within 1 kcal/mol from the corresponding global minimum. In some systems with flat potential energy surface, the full conformational space could be confined within that energy window. This will cause all the conformations of that isomer to be eliminated, and therefore its probability will be zero. To indicate that situation, the corresponding cells will be highlighted in gray (in the given example, column H, rows 13 and 14).
Ten_isomers sheets: the Excel file contains as many sheets as candidate structures are considered, labeled as “Tens_Isomer N”, where N is the isomer number. Each cell contains the isotropic shielding values corresponding to each ensemble (row) and Gaussian label (column). For example, the value shown in cell C2 (91.5338) is the isotropic shielding value of the atom nº 2 (according to Gaussian labeling scheme) computed using ensemble 2 [A-1-0-2].
Workflow and general recommendations
Step 1: Despite the new MESSI can handle any amount of isomers, keeping the number of candidates to a minimum has several advantages, as it reduces both the overall computational cost and the probability that the calculated data for an incorrect isomer ends up having better fit with the experimental values than the correct candidate.
Step 2: The conformational search should provide a good description of the conformational landscape of the system under study. Improper computational work might lead to potentially negative consequences in the overall results. Systematic sampling is always recommended, but impractical in highly flexible molecules. In those cases, stochastic searches using a reasonably large number of steps should be carried out. To avoid missing potentially relevant conformations, all conformations within a safe energy window from the corresponding global minimum should be kept. For this application, we recommend a 10 kcal/mol cutoff value using the MMFFaq force field.
Step 3: All conformations found in Step 2 must be fully optimized at the PCM/B3LYP/6-31G* level.
Step 4: After removing duplicates, all structures found must be submitted to NMR calculations at the level A (PCM/mPW1PW91/6-31+G**). In parallel, the same PCM/B3LYP/6-31G* optimized structures must be used as inputs for single point energy calculations at level B (SMD/B3LYP/6-31+G**). Important: MESSI requires that all conformations found to be considered, not just the most stable ones. Therefore, keeping only the most stable conformations found in Step 3 could give erroneous results. In the same way, it is important to respect the suggested theory levels, since MESSI was optimized for those levels.
Step 5: The output files must be compiled in a folder. Additionally, an Excel file with the experimental data and labels is needed. The NMR data must be assigned (know which shift corresponds to which nuclei). Using unassigned or misassigned NMR data can lead to erroneous results. The chemical shifts of equivalent nuclei that show fast interconversion should be averaged (such as the case of methyl groups, or some methylene groups). Treating the signal of each individual proton independently is wrong (for example, computing three different chemical shifts for the same methyl group). Another problem arises when dealing with diastereotopic methylene protons, which are ofen arbitrarily correlated. Unless the discrimination of both signals as pro-R and pro-S is made using additional NMR information (such as NOE or J coupling), the most convenient way to tackle this issue is to treat them as interchangeable signals.
Step 6: Run the script
messi.pyto perform the PCM-DP4+, SMD-DP4+ and MESSI probabilities calculations. The script opens a pop-up window that requests to select the folder that contains the Gaussian output files (either as*.log or *.out), and the Excel input file. The script feeds on the corresponding NMR and SCRF/SMD single point Gaussian output files. Both types of calculations could be run separately or together through the "link" option. The script automatically extracts the isotropic shielding values and energies from each output and classifies them per isomer. Finally, the chemical shifts are averaged according the filter type and correlated with the experimental data to use it in the DP4+ formalism. The results are printed in an Excel file named ‘MESSI_Results.xlsx’.
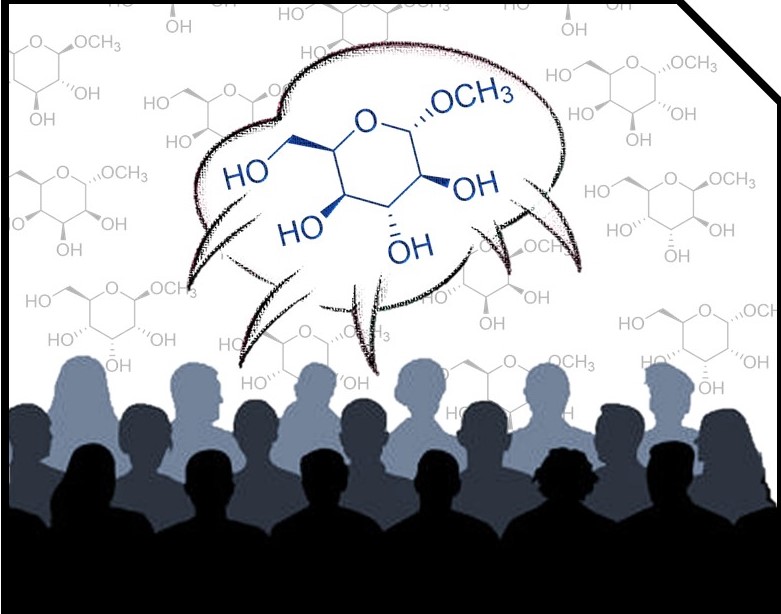
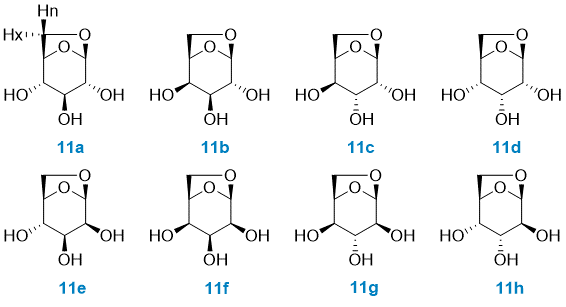
Case study: 1,6-anhydrohexopyranosides
In order to illustrate the MESSI workflow, we present the analysis of 1,6-anhydrohexopyranosides family. As indicated in the Figure, there are eight possible isomers.
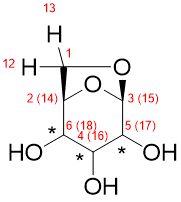
Following the recommended computational procedure, a total number of 130 conformers were found after the optimization at the B3LYP/6-31G* level (the standard for DP4+ calculations). Each structure was submitted to NMR and SCRF calculations at the PCM/mPW1PW91/6-31+G** and SMD/B3LYP/6-31+G** level respectively. The corresponding output files are provided in the Folder “Example”. According to Gaussian numbering scheme, the labels corresponding to each nuclei are given in Figure, Carbon label followed by its corresponding proton(s) label(s) between parenthesis.
MESSI Analysis
MESSI calculations were running for the eight possible distereoisomer, and placed in a folder. Once the script is run, the resulting excel report file “MESSI_Results” will be generated.
MESSI input and output excel files
Then execute messi_nmr and follow the steps.
Detail information is given in UserGuide.pdf included in the package. (Direct acces from GUI)
See the project repository for more details.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distributions
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file messi_nmr-0.1.14-py3-none-any.whl.
File metadata
- Download URL: messi_nmr-0.1.14-py3-none-any.whl
- Upload date:
- Size: 4.8 MB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.2 CPython/3.10.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
3032cd168d620bd1aa45a6e291ad9cf7dc6fc184a6d9d5a2ac1c20e364a299a4
|
|
| MD5 |
6d9ec86b03c7926d23b61d9d7d66457a
|
|
| BLAKE2b-256 |
e8948576f86c36dbc153f50f51f84556da94a193e5430575b1a561a9271dcbdd
|







