MetEvolSim (Metabolome Evolution Simulator) Python Package

Project description

Metabolome Evolution Simulator
A Python package to simulate the long-term evolution of metabolic levels.


MetEvolSim (Metabolome Evolution Simulator) is a Python package which provides numerical tools to simulate the long-term evolution of metabolic abundances in kinetic models of metabolic network. To use MetEvolSim, a SBML-formatted metabolic network model is required, along with kinetic parameters and initial metabolic concentrations. Additionally, the model must reach a stable steady-state, which is computed with Copasi software.
MetEvolSim is being developed by Charles Rocabert, Gábor Boross, Orsolya Liska and Balázs Papp.
If you are planning to use MetEvolSim for research or have encountered any issues with the software, do not hesitate to contact Charles Rocabert.


Table of contents
- Publications
- Dependencies
- Installation
- First usage
- Help
- Ready-to-use examples
- List of tested metabolic models
- Copyright
- License
Publications
- O. Liska, G. Boross, C. Rocabert, B. Szappanos, R. Tengölics, B. Papp. Principles of metabolome conservation in animals. Proceedings of the National Academy of Sciences 120 (35), e2302147120 (2023) (https://doi.org/10.1073/pnas.2302147120).
Dependencies
- Python ≥ 3,
- Numpy ≥ 1.21 (automatically installed when using pip),
- Python-libsbml ≥ 5.19 (automatically installed when using pip),
- NetworkX ≥ 2.6 (automatically installed when using pip),
- CopasiSE ≥ 4.27 (to be installed separately),
- pip ≥ 21.3.1 (optional).
Installation
• To install Copasi software, visit http://copasi.org/ and download the command line version CopasiSE.
• To install the latest release of MetEvolSim:
pip install MetEvolSim
Alternatively, download the latest release in the folder of your choice and unzip it. Then follow the instructions below:
# Navigate to the MetEvolSim folder
cd /path/to/MetEvolSim
# Install MetEvolSim Python package
python3 setup.py install
First usage
MetEvolSim has been tested with numerous publicly accessible metabolic networks; however, we cannot guarantee that it will be compatible with any model (please refer to the list of tested metabolic models).
The package includes the class Model to manipulate SBML models. Additionally, it is necessary to set up an objective function (a list of target reactions and their coefficients) and to provide the path of CopasiSE software. Please note that objective function coefficients are not operational in the current version of MetEvolSim.
# Import MetEvolSim package
import metevolsim
# Create an objective function
target_fluxes = [['ATPase', 1.0], ['PDC', 1.0]]
# Load the SBML metabolic model
model = metevolsim.Model(sbml_filename='glycolysis.xml',
objective_function=target_fluxes,
copasi_path='/Applications/COPASI/CopasiSE')
# Print some informations on the metabolic model
print(model.get_number_of_species())
print(model.get_wild_type_species_value('Glc'))
# Get a kinetic parameter at random
param = model.get_random_parameter()
print(param)
# Mutate this kinetic parameter with a log-scale mutation size 0.01
model.random_parameter_mutation(param, sigma=0.01)
# Compute wild-type and mutant steady-states
model.compute_wild_type_steady_state()
model.compute_mutant_steady_state()
# Run a metabolic control analysis on the wild-type
model.compute_wild_type_metabolic_control_analysis()
# This function will output two datasets:
# - output/wild_type_MCA_unscaled.txt containing unscaled control coefficients,
# - output/wild_type_MCA_scaled.txt containing scaled control coefficients.
# Compute all pairwise metabolite shortest paths
model.build_species_graph()
model.save_shortest_paths(filename="glycolysis_shortest_paths.txt")
# Compute a flux drop analysis to measure the contribution of each flux to the fitness
# (in this example, each flux is dropped at 1% of its original value)
model.flux_drop_analysis(drop_coefficient=0.01,
filename="flux_drop_analysis.txt",
owerwrite=True)
MetEvolSim offers two distinct numerical approaches for assessing the evolution of metabolic abundances:
- Evolution experiments, based on a Markov Chain Monte Carlo (MCMC) algorithm, Sensitivity analysis, that can either explore every kinetic parameter in a given range and record changes in associated fluxes and metabolic abundances (One-At-a-Time sensitivity analysis) or randomly explore the kinetic parameter space by randomly mutating a single kinetic parameter multiple times (random sensitivity analysis).
All numerical analysis output files are saved in the output subfolder.
Evolution experiments:
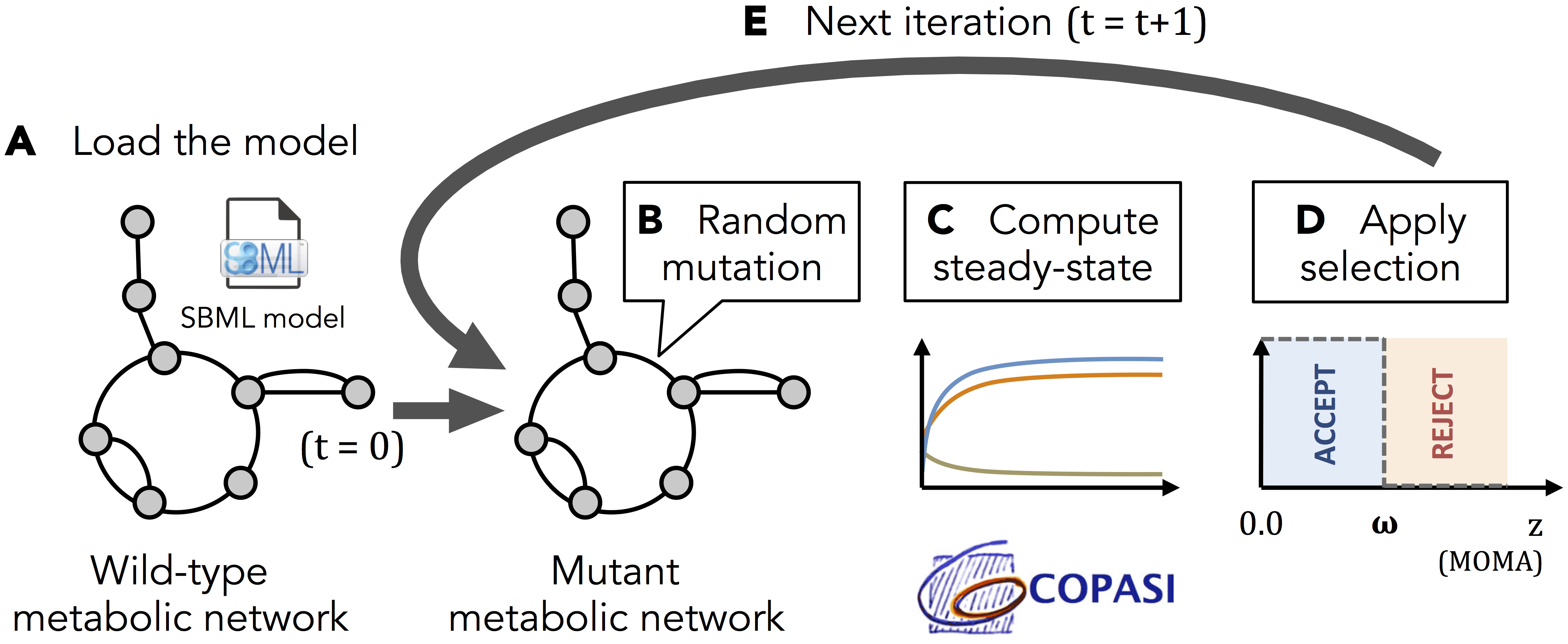
Algorithm overview: A. The model of interest is loaded as a wild-type from a SBML file (kinetic equations, kinetic parameter values and initial metabolic concentrations must be specified). B. At each iteration t, a single kinetic parameter is selected at random and mutated through a log10-normal distribution of standard deviation σ. C. The new steady-state is computed using Copasi software, and the MOMA distance z between the mutant and the wild-type target fluxes is computed. D. If z is under a given selection threshold ω, the mutation is accepted. Else, the mutation is discarded. E. A new iteration t+1 is computed.
There are six types of selection available:
MUTATION_ACCUMULATION: Run a mutation accumulation experiment by accepting all new mutations without any selection threshold,ABSOLUTE_METABOLIC_SUM_SELECTION: Run an evolution experiment by applying a stabilizing selection on the sum of absolute metabolic abundances,ABSOLUTE_TARGET_FLUXES_SELECTION: Run an evolution experiment by applying a stabilizing selection on the MOMA distance of absolute target fluxes,RELATIVE_TARGET_FLUXES_SELECTION: Run an evolution experiment by applying a stabilizing selection on the MOMA distance of relative target fluxes.
# Load a Markov Chain Monte Carlo (MCMC) instance
mcmc = metevolsim.MCMC(sbml_filename='glycolysis.xml',
objective_function=target_fluxes,
total_iterations=10000,
sigma=0.01,
selection_scheme="MUTATION_ACCUMULATION",
selection_threshold=1e-4,
copasi_path='/Applications/COPASI/CopasiSE')
# Initialize the MCMC instance
mcmc.initialize()
# Compute the successive iterations and write output files
stop_MCMC = False
while not stop_MCMC:
stop_mcmc = mcmc.iterate()
mcmc.write_output_file()
mcmc.write_statistics()
One-At-a-Time (OAT) sensitivity analysis:
For each kinetic parameter p, each metabolic abundance [Xi] and each flux νj, the algorithm numerically computes relative derivatives and control coefficients.
# Load a sensitivity analysis instance
sa = metevolsim.SensitivityAnalysis(sbml_filename='glycolysis.xml',
copasi_path='/Applications/COPASI/CopasiSE')
# Run the full OAT sensitivity analysis
sa.run_OAT_analysis(factor_range=1.0, factor_step=0.01)
Random sensitivity analysis:
At each iteration, a single kinetic parameter p is mutated at random in a log10-normal distribution of size σ, and relative derivatives and control coefficients are computed.
# Load a sensitivity analysis instance
sa = metevolsim.SensitivityAnalysis(sbml_filename='glycolysis.xml',
copasi_path='/Applications/COPASI/CopasiSE')
# Run the full OAT sensitivity analysis
sa.run_random_analysis(sigma=0.01, nb_iterations=1000)
Help
To get assistance with a MetEvolSim class or method, use the Python help function:
help(metevolsim.Model.set_species_initial_value)
To get a brief overview and a list of parameters and outputs:
Help on function set_species_initial_value in module metevolsim:
set_species_initial_value(self, species_id, value)
Set the initial concentration of the species 'species_id' in the
mutant model.
Parameters
----------
species_id: str
Species identifier (as defined in the SBML model).
value: float >= 0.0
Species abundance.
Returns
-------
None
(END)
Ready-to-use examples
Ready-to-use examples come pre-packaged with MetEvolSim package. They can also be downloaded here: https://github.com/charlesrocabert/MetEvolSim/raw/master/example/example.zip.
List of tested metabolic models
| Reference | Model | Running with MetEvolSim |
|---|---|---|
| Bakker et al. (1997) | Trypanosoma brucei glycolysis | :x: |
| Curto et al. (1998) | Human purine metabolism | :x: |
| Mulquiney et al. (1999) | Human erythrocyte | :white_check_mark: |
| Jamshidi et al. (2001) | Red blood cell | :x: |
| Bali et al. (2001) | Red blood cell glycolysis | :white_check_mark: |
| Lambeth et al. (2002) | Skeletal muscle glycogenolysis | :white_check_mark: |
| Holzhutter et al. (2004) | Human erythrocyte | :white_check_mark: |
| Beard et al. (2005) | Mitochondrial respiration | :x: |
| Banaji et al. (2005) | Cerebral blood flood control | :white_check_mark: |
| Bertram et al. (2006) | Mitochondrial ATP production | :x: |
| Bruck et al. (2008) | Yeast glycolysis | :white_check_mark: |
| Reed et al. (2008) | Glutathione metabolism | :x: |
| Curien et al. (2009) | Aspartame metabolism | :x: |
| Jerby et al. (2010) | Human liver metabolism | :x: |
| Li et al. (2010) | Yeast glycolysis | :x: |
| Bekaert et al. (2010) | Mouse metabolism reconstruction | :x: |
| Bordbar et al. (2011) | Human multi-tissues | :x: |
| Koenig et al. (2012) | Hepatocyte glucose metabolism | :white_check_mark: |
| Messiha et al. (2013) | Yeast glycolysis + pentose phosphate | :white_check_mark: |
| Mitchell et al. (2013) | Liver iron metabolism | :x: |
| Stanford et al. (2013) | Yeast whole cell model | :x: |
| Bordbar et al. (2015) | Red blood cell | :x: |
| Costa et al. (2016) | E. coli core metabolism | :white_check_mark: |
| Millard et al. (2016) | E. coli core metabolism | :white_check_mark: |
| Bulik et al. (2016) | Hepatic glucose metabolism | :white_check_mark: |
Copyright
Copyright © 2018-2023 Charles Rocabert, Gábor Boross, Orsolya Liska and Balázs Papp. All rights reserved.
License
This program is free software: you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation, either version 3 of the License, or (at your option) any later version.
This program is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU General Public License for more details.
You should have received a copy of the GNU General Public License along with this program. If not, see http://www.gnu.org/licenses/.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file MetEvolSim-1.0.0.tar.gz.
File metadata
- Download URL: MetEvolSim-1.0.0.tar.gz
- Upload date:
- Size: 58.7 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.2 CPython/3.11.5
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
ab9513750aedd3010ca0697cd27e4204b64430037ef938b34269d201d94c0fb9
|
|
| MD5 |
df451cc69acb95627052149aaec4d5f3
|
|
| BLAKE2b-256 |
2cb06c4e91d8b30dce0a186b75bd7bd6f0c3bb1c0f7a0715eb724b60dc00f4fd
|
File details
Details for the file MetEvolSim-1.0.0-py3-none-any.whl.
File metadata
- Download URL: MetEvolSim-1.0.0-py3-none-any.whl
- Upload date:
- Size: 35.1 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.2 CPython/3.11.5
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
a1c6b7f409d0bf965e6fe44c732501271ac631f1f8828f816f82e09db6234476
|
|
| MD5 |
3c3d1b32eaa4231f446b755c169e4b4a
|
|
| BLAKE2b-256 |
90e34791c5e5997bb90aa279928121d8b7db871f50ca4cb32fe442203ffb015e
|







