Identification of allele-specific events in sequencing experiments.

Project description

MixALime: Mixture models for Allelic Imbalance Estimation
MixALime is a tool for the identification of allele-specific variants (ASVs or allele-specific events, ASEs) in high-throughput sequencing data. It works by modelling counts data as a mixture of two Negative Binomial or Beta Negative Binomial distributions (where the latter is more applicable in case of noisy data at a cost of loss of sensitivity).
Homepage & Docs
MixALime features and usage guidelines are best explained at the project's homepage: mixalime.georgy.top
General workflow
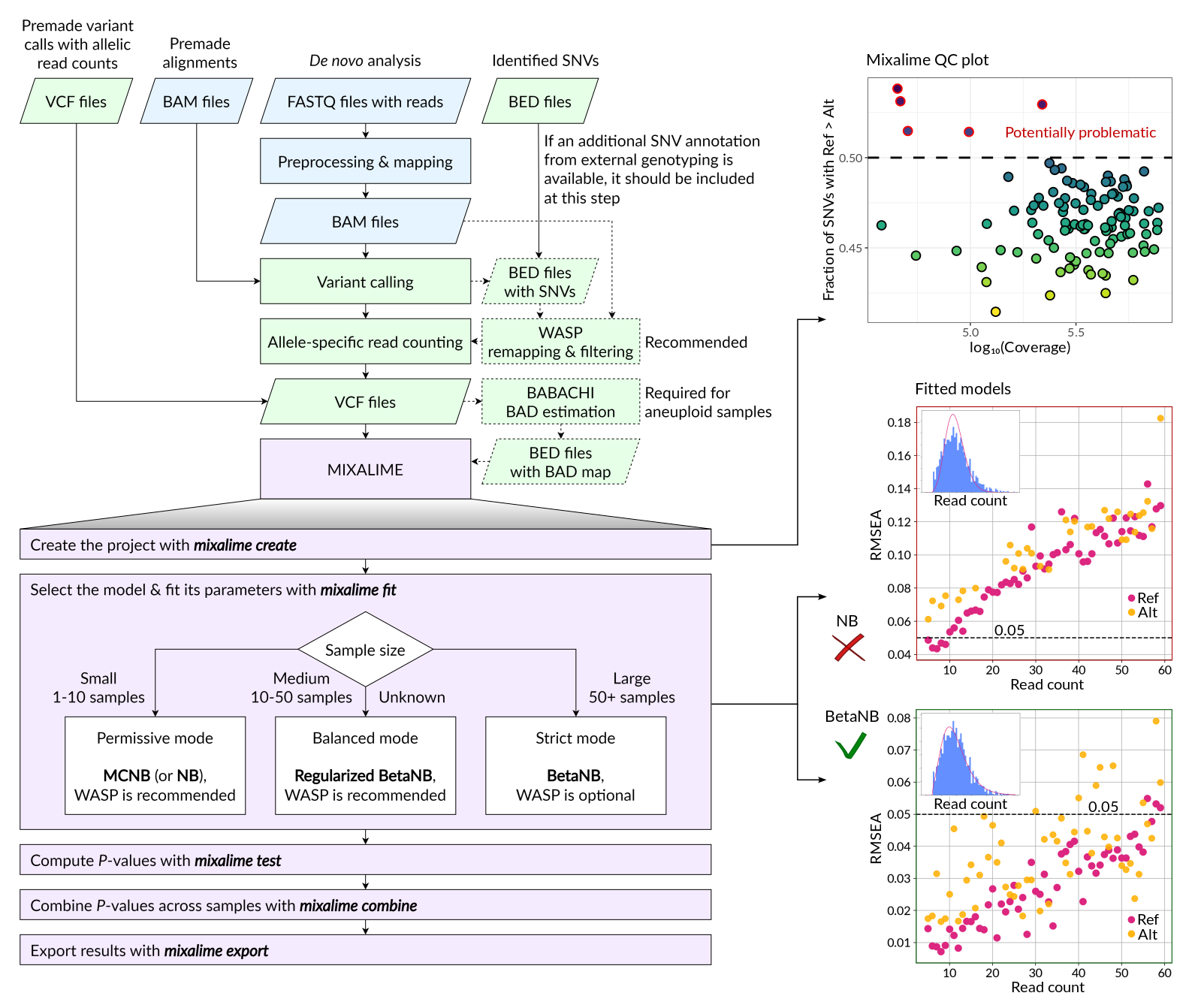
MixALime allows direct ASV (allele-specific variants) estimation from the VCF files generated by a variant caller from BAM alignments. To obtain the input files for MixALime, the users may start from FASTQ files, align them to the genome, and perform variant calling or use premade alignment files to call SNVs or the existing premade VCF files. In former cases, the WASP remapping and filtering procedure may be additionally performed to improve the ASV calling, especially for low sample sizes. If the external genotyping information is available it can be used to expand or filter the SNV calls. With the VCF files, BABACHI can be used to reconstruct the BAD maps for aneuploid samples before running MixALime. Finally, ASVs can be estimated with the MixALime using the model chosen according to the sample size (see also Figure 3D). Given the genotype calls are reliable (e.g. available from external data or called from a large collection of samples), the default MixALime filter requiring at least 5 reads at both alleles can be relaxed or omitted. As a result, MixALime will produce several QC plots together with the ASV tables. The former include sample-level reference bias estimations which can be used to detect outliers with extreme reference bias (unknown technical issues with the original data or incorrectly assigned genotypes, top right), as well as the goodness-of-fit assessment plots (middle and bottom right).
System requirements
OS
In our work, we mostly used Ubuntu 20.04.6 LTS (GNU/Linux 5.15.0-82-generic x86_64) and Manjaro 6.1.62-1. We never tested MixALime on Windows systems.
Software dependencies
MixALime is best used with Python 3.9. Package dependencies that could affect MixALime results are the JAX 0.4.20, scipy 1.11.3 and numpy 1.26.2.
Hardware requirements
It is best to use MixALime with plenty of RAM available if one wishes to use the parallelization to the full extent. In that case, make sure that your system has at least 16GB of RAM. However, the RAM requirement is depends on a dataset.
Input requirements
Mixalime expects input VCF/BED files to contain biallelic SNPs with Ref alleles corresponding to the reference genome. Multiallelic SNPs can be split into biallelic records with bcftools norm --multiallelics, Ref alleles can be checked with —check-ref bcftools option.
Installation guide
MixALime can be easily installed with the pip package manager.
> pip3 install mixalime
Alternatively, MixALime can be installed from the git repository:
> git clone https://github.com/autosome-ru/MixALime
> cd MixALime
> python3 setup.py install
Regardless of the chosen method, installation time of the MixALime alone is capped by the internet connection speed, but should take no longer than a minute, granted that all the dependency packages are already installed.
Instructions for use
The package is almost easy to use and we advise everyone to just jump straight to installing MixALime and invoking the help command in a command line:
> pip3 install mixalime
> mixalime --help
We believe that the help section of MixALime covers its functionality well enough. Furthermore, the package arrives with a small demo dataset included and an easy-to-follow instruction in the abovementioned help section. Furthermore, note that all commands avaliable in MixALime's command-line interface have their own help page too, e.g.:
> mixalime fit --help
So do not waste your time looking for how-to-clues or tutorials here, just use --help.
Yet, for the sake of following the social norms that impose a requirement of README files to be useful, in the next section you'll find the excerpt from --help command as well as some other possibly useful details.
Demo
A typical MixALime session consists of sequential runs of create, fit, test, combine and, finally, export all, plot commands. For instance, we provide a demo dataset that consists of a bunch of BED-like files with allele counts at SNVs (just for the record, MixALime can work with most vcf and BED-like file formats):
> mixalime export demo
A scorefiles folder should appear now in a working directory with a plenty of BED-like files. First, we'd like to parse those files into a MixALime-friendly and efficient data structures for further usage, as well as perform some basic filtering if necessary:
> mixalime create myprojectname scorefiles --no-snp-bad-check
Expected output:
┏━━━━━━┳━━━━━━━┳━━━━━━━━━━━━━━━┓
┃ BAD ┃ SNVs ┃ Obvservations ┃
┡━━━━━━╇━━━━━━━╇━━━━━━━━━━━━━━━┩
│ 1.00 │ 10320 │ 24893 │
│ 1.33 │ 7408 │ 13359 │
│ 1.50 │ 12409 │ 37584 │
│ 2.00 │ 41763 │ 199208 │
│ 2.50 │ 2195 │ 3585 │
│ 3.00 │ 6897 │ 26558 │
│ 4.00 │ 1677 │ 2420 │
│ 5.00 │ 712 │ 926 │
│ 6.00 │ 731 │ 1143 │
└──────┴───────┴───────────────┘
Total unique SNVs: 84112, total observations: 309676
✔️ Done! time: 9.72 s.
By default, MixALime throws an error if the same SNV belongs regions of different background allelic dosage (BAD) in data, as it is not possible in a single cell type. However, for datasets of multiple samples, this is fully OK, hence we turn this check off with the --no-snp-bad-check. The output:
Then we fit model parameters to the data with Negative Binomial distribution:
> mixalime fit myprojectname NB
Expected output:
WARNING:root:Total number of samples at BAD 6.0 is less than a window size (1143 < 10000). Number of samples is too small for a sensible fit, a conservative fit will be used.
WARNING:root:Total number of samples at BAD 2.5 is less than a window size (3585 < 10000). Number of samples is too small for a sensible fit, a conservative fit will be used.
WARNING:root:Total number of samples at BAD 5.0 is less than a window size (926 < 10000). Number of samples is too small for a sensible fit, a conservative fit will be used.
WARNING:root:Total number of samples at BAD 4.0 is less than a window size (2420 < 10000). Number of samples is too small for a sensible fit, a conservative fit will be used.
✔️ Done! time: 38.21 s.
Warnings are fine. More on them in the tutorial. Next we obtain raw p-values:
> mixalime test myprojectname
Expected output:
✔️ Done! time: 47.96 s.
Usually we'd want to combine p-values across samples and apply a FDR correction:
> mixalime combine myprojectname
Expected output:
┏━━━━━━┳━━━━━━┳━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━━━━━━━┓
┃ ┃ ┃ ┃ Total significant ┃
┃ Ref ┃ Alt ┃ Both ┃ (Percentage of total SNVs) ┃
┡━━━━━━╇━━━━━━╇━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━━━━━━━┩
│ 1325 │ 1127 │ 1 │ 2451 (7.28%) │
└──────┴──────┴──────┴────────────────────────────┘
Total SNVs tested: 33689
✔️ Done! time: 10.99 s.
Finally, we obtain fancy plots fir diagnostic purposes and easy-to-work-with tabular data:
> mixalime export all myprojectname results_folder
> mixalime plot myprojectname results_folder
Expected output:
✔️ Done! time: 4.29 s.
✔️ Done! time: 28.00 s.
You'll find everything of interest in results_folder.
Combination of p-values across groups
Note: a popular synonym for "combination" in this context is aggregation.
One important feature that is not covered by the glorified --help in a very obvious fashion is a combination of p-values across separate groups (e.g. one group can be a treatment and the other is a control). The combine command with default options combines all the p-values. This can be changed by supplying the --group option followed by either a list of filenames that make up that group or a file that contains a list (newline-separated) of those files (the most convenient approach, probably), e.g.:
> mixalime combine --subname treatment -g vcfs/file1.vcf.gz -g vfcfs/file2.vfc.gz -g vcfs/file3.vcf.gz myproject
> mixalime combine --subname control -g vcfs/file4.vcf.gz -g vfcfs/file5.vfc.gz -g vcfs/file6.vcf.gz myproject
or
> mixalime combine --subname treatment -g group_treatment.tsv combine myproject
> mixalime combine --subname control -g group_control.tsv combine myproject
The --subname option is necessary if you wish to avoid different combine invocations overwriting each other.
Scoring models
The package provides a variety of models for datasets of varying dispersion:
| Name | Dataset variance | Comments |
|---|---|---|
| NB | Low | Fastest parameter estimation; might be too liberal for some datasets |
| MCNB | Medium-low | Marginalized Compound Negative Binomial (MCNB), the safest compromise between liberal NB and conservative BetaNB |
| BetaNB | High | Introduces an extra parameter to control for higher variance, fits most datasets perfectly, yet the scoring is often overly conservative |
| Regularized BetaNB | Depends | Introduces penalty on the extra parameter to make the model less likely to overfit with the --regul-a command. Requires tuning the regularization hyperparameter alpha which might not be feasible |
The name of the appropriate model is supplied to the fit command as an argument (except for regularized BetaNB which is just an fit ProjectName BetaNB with an --regul-a alpha_value option where alpha_value is your hyperparameter value, e.g. 1.0).
Binomial and beta-binomial models
MixALime also can do good old-fashion binomial and beta-binomial tests. They can be done with the separate test_binom (with --beta flag if you want beta-binomial). Note, that with this command you can skip the fit (as not fit is done here, except for beta-binomial, where a single variance parameter is estimated for each BAD) and test step.
Inner clockworks & Citing
For the time being, you can cite our technical arXiv paper that explains MixALime's inner clockworks in a great detail:
@misc{mixalimeMath,
title={MIXALIME: MIXture models for ALlelic IMbalance Estimation in high-throughput sequencing data},
author={Georgy Meshcheryakov and Sergey Abramov and Aleksandr Boytsov and Andrey I. Buyan and Vsevolod J. Makeev and Ivan V. Kulakovskiy},
year={2023},
eprint={2306.08287},
archivePrefix={arXiv},
primaryClass={stat.AP},
doi = {10.48550/arXiv.2306.08287},
url = {https://doi.org/10.48550/arXiv.2306.08287}
}
and the practical NatComm paper:
@article{mixalimeUsage,
title = {Statistical framework for calling allelic imbalance in high-throughput sequencing data},
volume = {16},
ISSN = {2041-1723},
url = {http://dx.doi.org/10.1038/s41467-024-55513-2},
DOI = {10.1038/s41467-024-55513-2},
number = {1},
journal = {Nature Communications},
publisher = {Springer Science and Business Media LLC},
author = {Buyan, Andrey and Meshcheryakov, Georgy and Safronov, Viacheslav and Abramov, Sergey and Boytsov, Alexandr and Nozdrin, Vladimir and Baulin, Eugene F. and Kolmykov, Semyon and Vierstra, Jeff and Kolpakov, Fedor and Makeev, Vsevolod J. and Kulakovskiy, Ivan V.},
year = {2025},
month = feb
}
License
MixALime is distributed under WTFPL. If you prefer more standard licenses, feel free to treat WTFPL as CC-BY.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file mixalime-2.29.3.tar.gz.
File metadata
- Download URL: mixalime-2.29.3.tar.gz
- Upload date:
- Size: 5.5 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.12.8
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
3441b81eb7040406376f5963456d8fe694355d6046d66879c9a35fd5c9e51b39
|
|
| MD5 |
ef3c6f9a1e6ae1b9f3ea99899931f360
|
|
| BLAKE2b-256 |
e6d23dde6725cfcc33ea6c0be54ee42800290cbe5e4175ae7a9d9acd1dc73ef8
|







