Microbe-metabolite interactions through neural networks

Project description

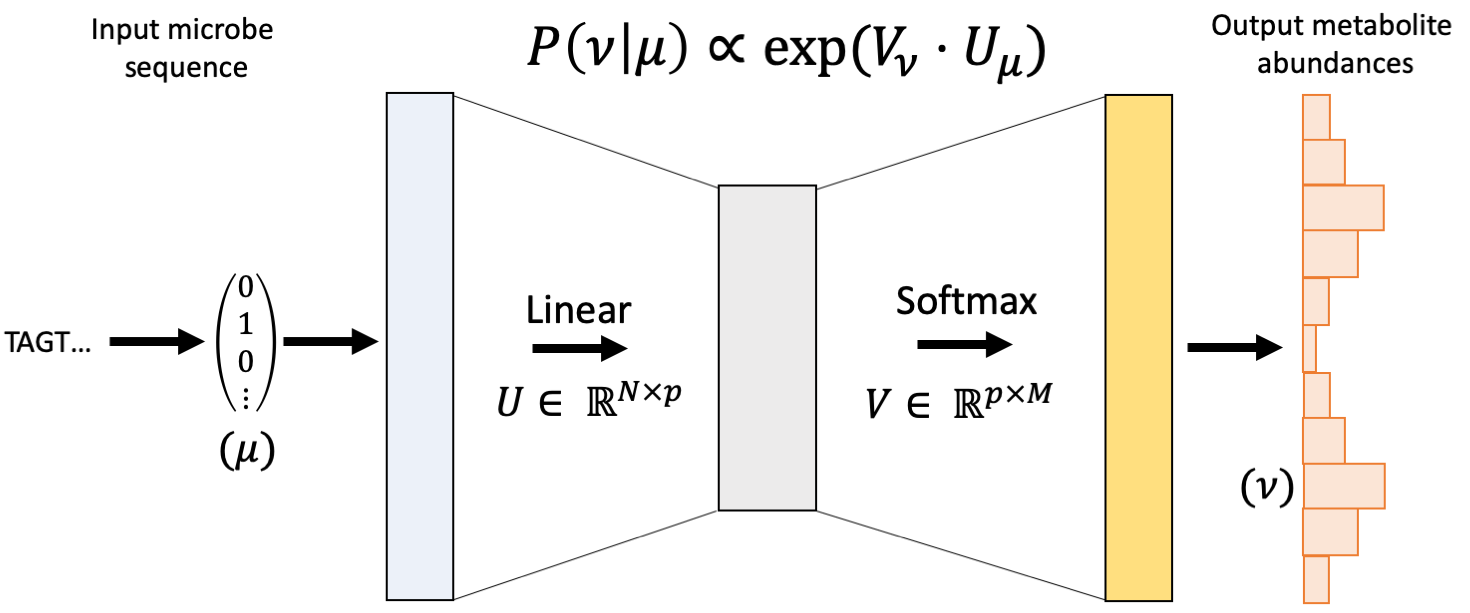
mmvec
Neural networks for estimating microbe-metabolite interactions through their co-occurence probabilities.
Installation
MMvec can be installed via pypi as follows
pip install mmvec
If you are planning on using GPUs, be sure to pip install tensorflow-gpu <= 1.14.0.
MMvec can also be installed via conda as follows
conda install mmvec -c conda-forge
Note that this option may not work in cluster environments, it maybe workwhile to pip install within a virtual environment. It is possible to pip install mmvec within a conda environment, including qiime2 conda environments. However, pip and conda are known to have compatibility issues, so proceed with caution.
Finally, MMvec is only compatible with qiime2 environments 2020.6 or before. Stay tuned for future updates.
Input data
The two basic tables required to run mmvec are:
- Metabolite counts (.biom): A table with metabolites in rows and samples in columns.
- Microbe abundance (.biom): A relative abundance table with microbial species in rows and samples in columns.
Getting started
To get started you can run a quick example as follows. This will learn microbe-metabolite vectors (mmvec) which can be used to estimate microbe-metabolite conditional probabilities that are accurate up to rank.
mmvec paired-omics \
--microbe-file examples/cf/otus_nt.biom \
--metabolite-file examples/cf/lcms_nt.biom \
--summary-dir summary
While this is running, you can open up another session and run tensorboard --logdir . for diagnosis, see FAQs below for more details.
If you investigate the summary folder, you will notice that there are a number of files deposited.
See the following url for a more complete tutorial with real datasets.
https://github.com/knightlab-analyses/multiomic-cooccurences
More information can found under mmvec --help
Qiime2 plugin
If you want to run this in a qiime environment, install this in your qiime2 conda environment (see qiime2 installation instructions here) and run the following
pip install git+https://github.com/biocore/mmvec.git
qiime dev refresh-cache
This should allow your q2 environment to recognize mmvec. Before we test
the qiime2 plugin, go to the examples/cf folder and run the following commands to import an example dataset
qiime tools import \
--input-path otus_nt.biom \
--output-path otus_nt.qza \
--type FeatureTable[Frequency]
qiime tools import \
--input-path lcms_nt.biom \
--output-path lcms_nt.qza \
--type FeatureTable[Frequency]
Then you can run mmvec
qiime mmvec paired-omics \
--i-microbes otus_nt.qza \
--i-metabolites lcms_nt.qza \
--p-summary-interval 1 \
--output-dir model_summary
In the results, there are three files, namely model_summary/conditional_biplot.qza, model_summary/conditionals.qza and model_summary/model_stats.qza.
The conditional biplot is a biplot representation the
conditional probability matrix so that you can visualize these microbe-metabolite interactions in an exploratory manner. This can be directly visualized in
Emperor as shown below. We also have the estimated conditional probability matrix given in results/conditionals.qza,
which an be unzip to yield a tab-delimited table via unzip results/conditionals. Each row can be ranked,
so the top most occurring metabolites for a given microbe can be obtained by identifying the highest co-occurrence probabilities for each microbe.
These log conditional probabilities can also be viewed directly with qiime metadata tabulate. This can be
created as follows
qiime metadata tabulate \
--m-input-file results/conditionals.qza \
--o-visualization conditionals-viz.qzv
Then you can run the following to generate a emperor biplot.
qiime emperor biplot \
--i-biplot conditional_biplot.qza \
--m-sample-metadata-file metabolite-metadata.txt \
--m-feature-metadata-file taxonomy.tsv \
--o-visualization emperor.qzv
The resulting biplot should look like something as follows
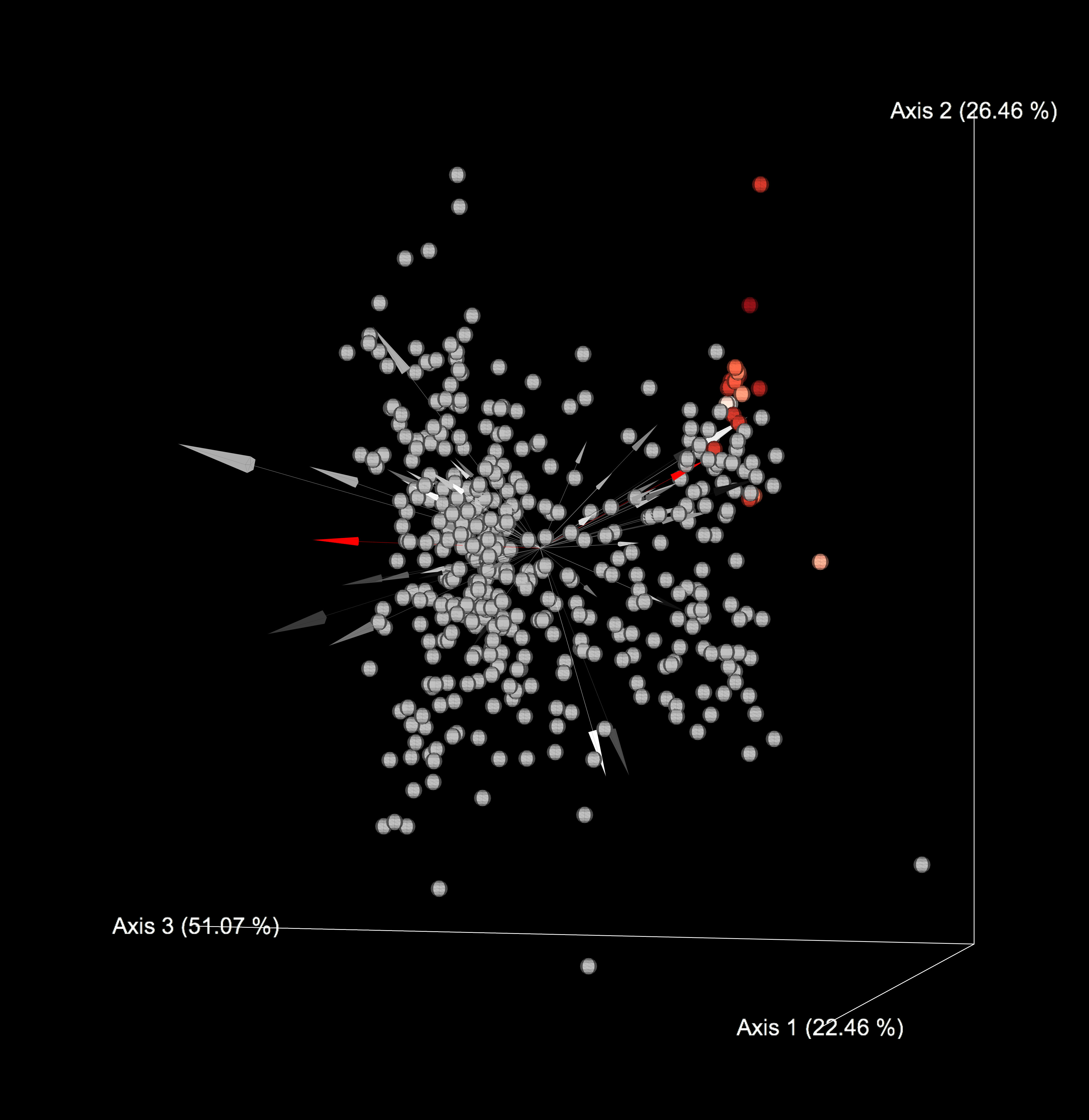
Here, the metabolite represent points and the arrows represent microbes. The points close together are indicative of metabolites that frequently co-occur with each other. Furthermore, arrows that have a small angle between them are indicative of microbes that co-occur with each other. Arrows that point in the same direction as the metabolites are indicative of microbe-metabolite co-occurrences. In the biplot above, the red arrows correspond to Pseudomonas aeruginosa, and the red points correspond to Rhamnolipids that are likely produced by Pseudomonas aeruginosa.
Another way to examine these associations is to build heatmaps of the log
conditional probabilities between observations, using the heatmap action:
qiime mmvec heatmap \
--i-ranks ranks.qza \
--m-microbe-metadata-file taxonomy.tsv \
--m-microbe-metadata-column Taxon \
--m-metabolite-metadata-file metabolite-metadata.txt \
--m-metabolite-metadata-column Compound_Source \
--p-level 5 \
--o-visualization ranks-heatmap.qzv
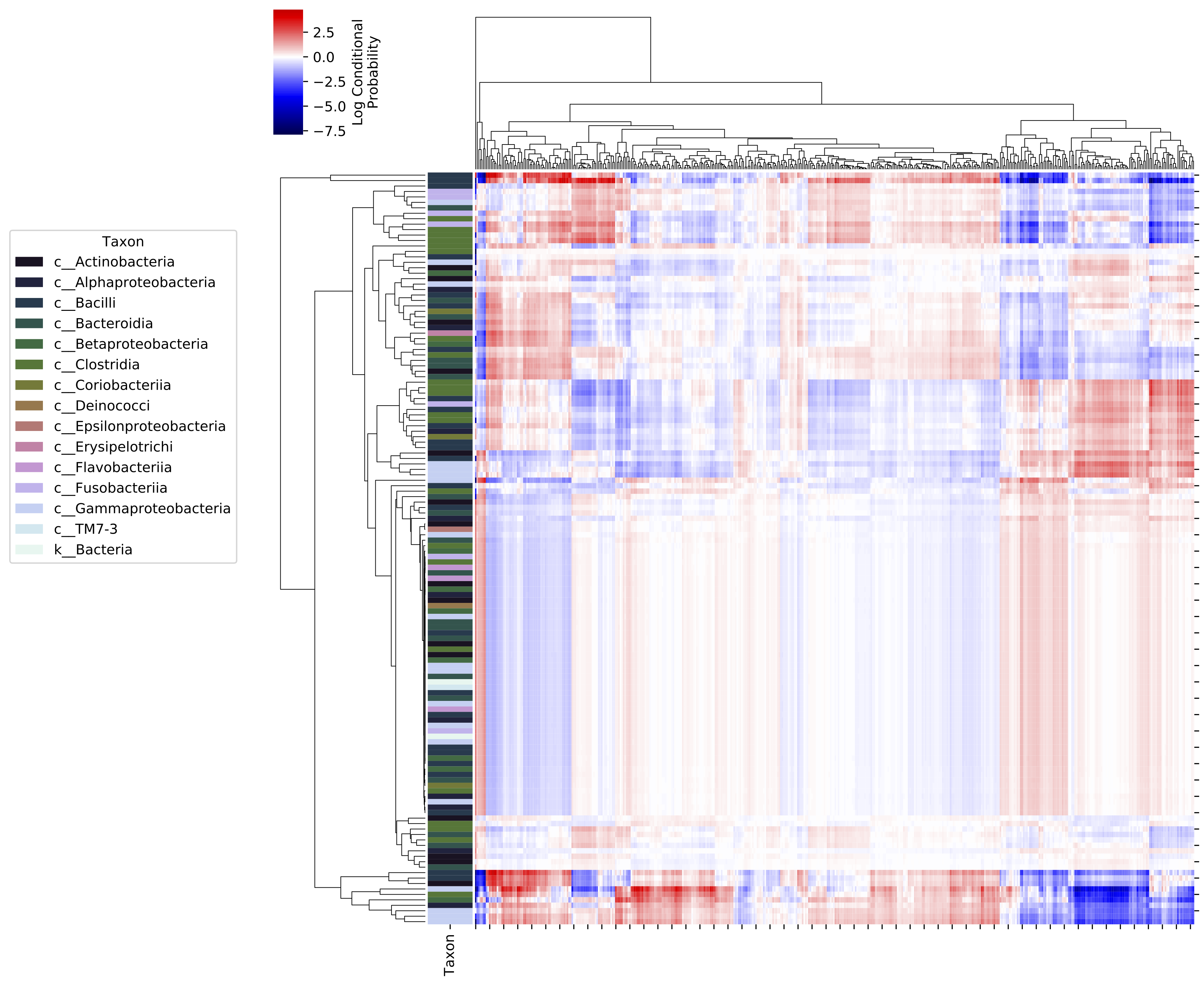
This action generates a clustered heatmap displaying the log conditional
probabilities between microbes and metabolites. Larger positive log conditional
probabilities indicate a stronger likelihood of co-occurrence. Low and negative
values indicate no relationship, not necessarily a negative correlation. Rows
(microbial features) can be annotated according to feature metadata, as shown
in this example; we provide a taxonomic classification file and the semicolon-
delimited taxonomic rank (level) that should be displayed in the color-coded
margin annotation. Set level to -1 to display the full annotation
(including of non-delimited feature metadata). Separate parameters are
available to annotate the x-axis (metabolites) in a similar fashion. Row and
column clustering can be adjusted using the method and metric parameters.
This action will generate a heatmap that looks similar to this:
Biplots and heatmaps give a great overview of co-occurrence associations, but
do not provide information about the abundances of these co-occurring features
in each sample. This can be done with the paired-heatmap action:
qiime mmvec paired-heatmap \
--i-ranks ranks.qza \
--i-microbes-table otus_nt.qza \
--i-metabolites-table lcms_nt.qza \
--m-microbe-metadata-file taxonomy.tsv \
--m-microbe-metadata-column Taxon \
--p-features TACGAAGGGTGCAAGCGTTAATCGGAATTACTGGGCGTAAAGCGCGCGTAGGTGGTTCAGCAAGTTGGATGTGAAATCCCCGGGCTCAACCTGGGAACTGCATCCAAAACTACTGAGCTAGAGTACGGTAGAGGGTGGTGGAATTTCCTG \
--p-features TACGTAGGTCCCGAGCGTTGTCCGGATTTATTGGGCGTAAAGCGAGCGCAGGCGGTTAGATAAGTCTGAAGTTAAAGGCTGTGGCTTAACCATAGTAGGCTTTGGAAACTGTTTAACTTGAGTGCAAGAGGGGAGAGTGGAATTCCATGT \
--p-top-k-microbes 0 \
--p-normalize rel_row \
--p-top-k-metabolites 100 \
--p-level 6 \
--o-visualization paired-heatmap-top2.qzv
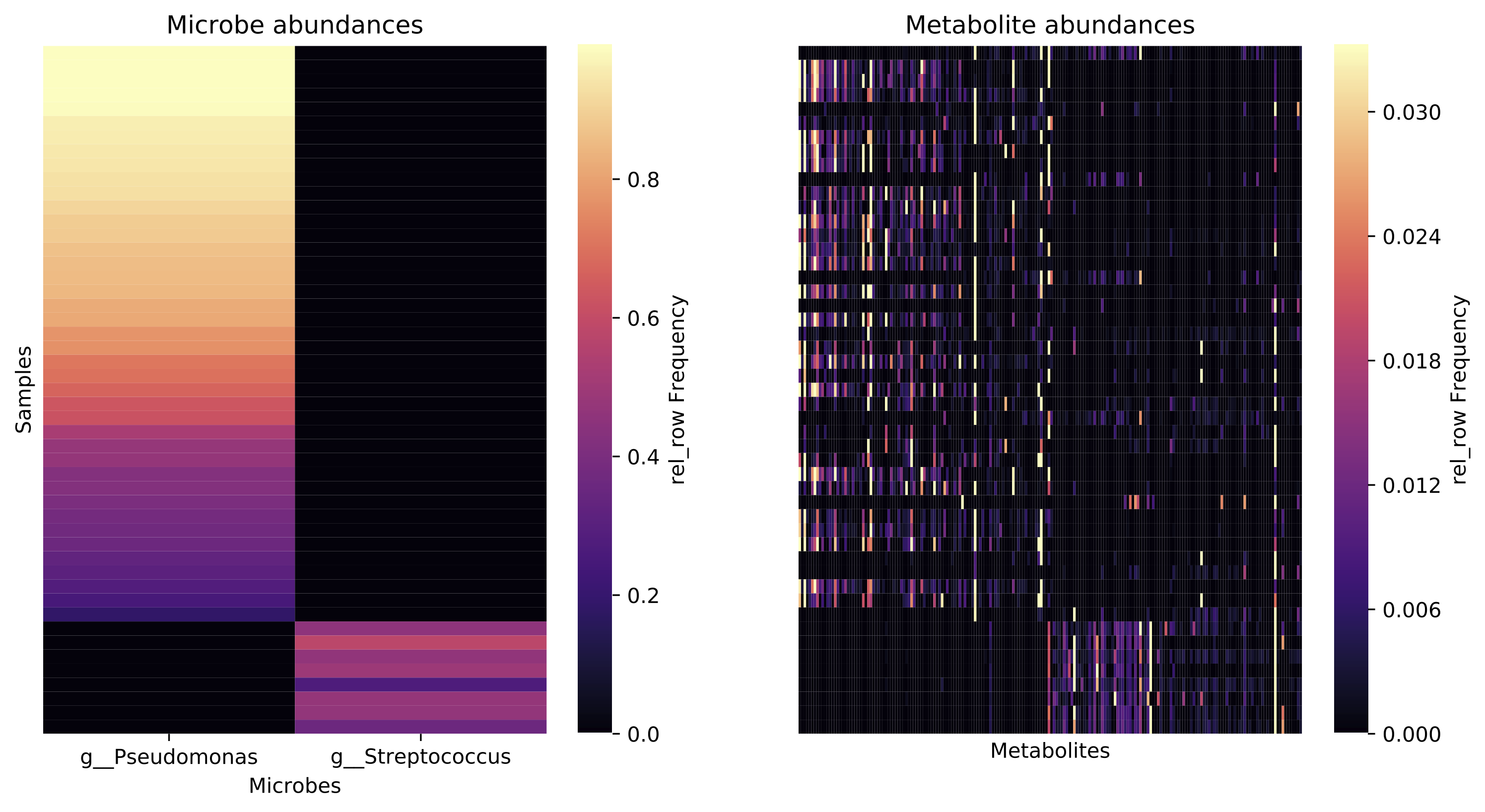
This action generates paired heatmaps that are aligned on the y-axis (sample
IDs): the left panel displays the abundances of each selected microbial feature
in each sample, and the right panel displays the abundances of the top k
metabolite features associated with each of these microbes in each sample.
Microbes can be selected automatically using the top-k-microbes parameter
(which selects the microbes with the top k highest relative abundances) or they
can be selected by name using the features parameter (if using the QIIME 2
plugin command-line interface as shown in this example, multiple features are
selected by passing this parameter multiple times, e.g., --p-features feature1 --p-features feature2; for python interfaces, pass a list of features:
features=[feature1, feature2]). As with the heatmap action, microbial
features can be annotated by passing in microbe-metadata and specifying a
taxonomic level to display. The output looks something like this:
More information behind the actions and parameters can found under qiime mmvec --help
Model diagnostics
QIIME2 Convergence Summaries
If you are using the qiime2 interface, there won't be a tensorboard interface. But there will still be training loss curves and cross-validation statistics reported, which are currently not available in the tensorboard interface. To run this with a single model, run the following
qiime mmvec summarize-single \
--i-model-stats model_summary/model_stats.qza \
--o-visualization model-summary.qzv
An example of what this will look like is given as follows
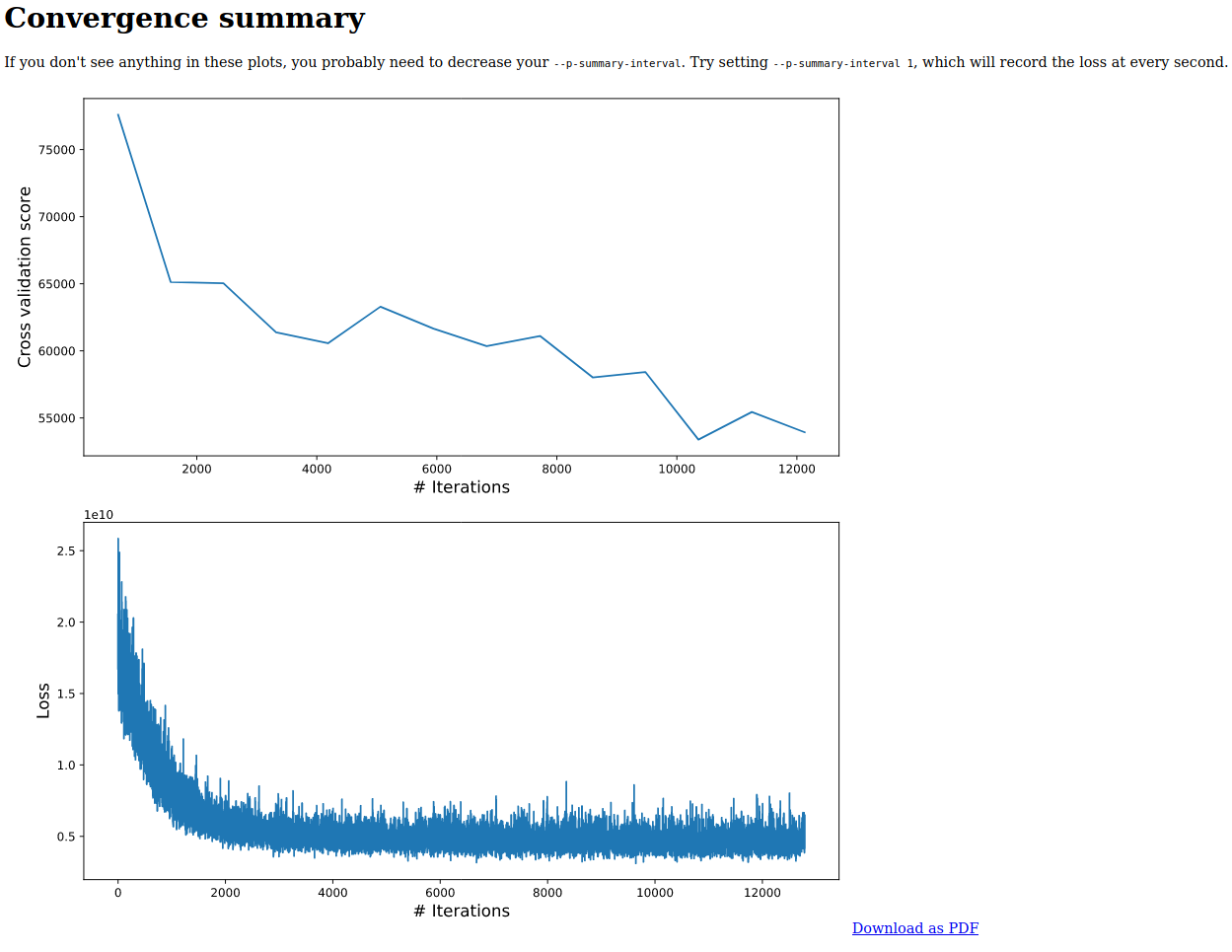
Null models and QIIME 2 + MMvec
If you're running Songbird through QIIME 2, the
qiime mmvec summarize-paired command allows you to view two sets of
diagnostic plots at once as follows:
# Null model with only biases
qiime mmvec paired-omics \
--i-microbes otus_nt.qza \
--i-metabolites lcms_nt.qza \
--p-latent-dim 0 \
--p-summary-interval 1 \
--output-dir null_summary
qiime mmvec summarize-paired \
--i-model-stats model_summary/model_stats.qza \
--i-baseline-stats null_summary/model_stats.qza \
--o-visualization paired-summary.qzv
An example of what this will look like is given as follows
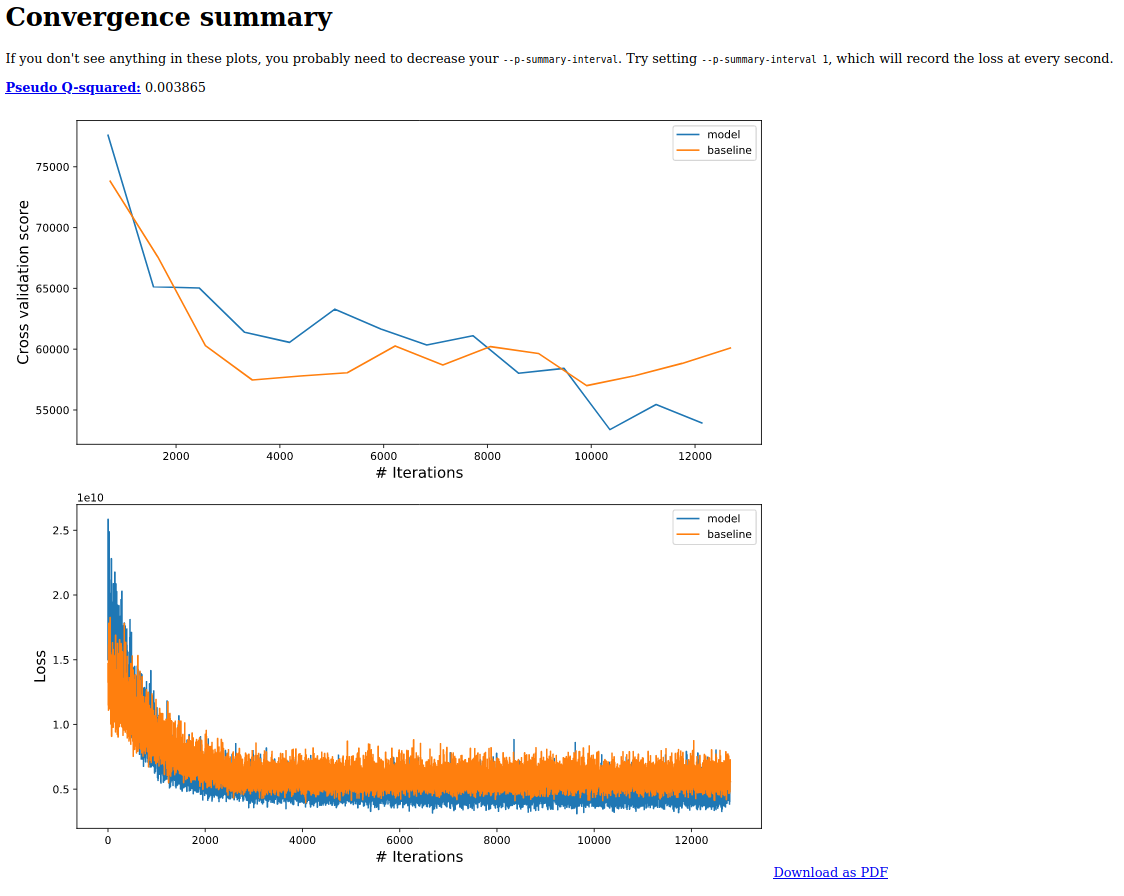
It is important to note here that the null model has a worst cross-validation error than the first MMvec model we trained. However to make the models exactly comparable, the same samples must be used for training and cross-validation. See the --p-training-column option to manually specify samples for training and testing.
These summaries can also be extended to analyze any two models of interest. This can help with picking optimal hyper-parameters.
Interpreting Q2 values
The Q2 score is adapted from the Partial least squares literature. Here it is given by Q^2 = 1 - m1/m2, where m1 indicates the average absolute model error and m2 indicates the average absolute null or baseline model error. If Q2 is close to 1, that indicates a high predictive accuracy on the cross validation samples. If Q2 is low or below zero, that indicates poor predictive accuracy, suggesting possible overfitting. This statistic behaves similarly to the R2 classically used in a ordinary linear regression if --p-formula is "1" in the m2 model.
If the Q2 score is extremely close to 0 (or negative), this indicates that the model is overfit or that the metadata supplied to the model are not predictive of microbial composition across samples. You can think about this in terms of "how does using the metadata columns in my formula improve a model?" If there isn't really an improvement, then you may want to reconsider your formula.
... But as long as your Q2 score is above zero, your model is learning something useful.
FAQs
Q: Looks like there are two different commands, a standalone script and a qiime2 interface. Which one should I use?!?
A: It'll depend on how deep in the weeds you'll want to get. For most intents and purposes, the qiime2 interface will more practical for most analyses. There are 3 major reasons why the standalone scripts are more preferable to the qiime2 interface, namely
-
Customized acceleration : If you want to bring down your runtime from a few days to a few hours, you may need to compile Tensorflow to handle hardware specific instructions (i.e. GPUs / SIMD instructions). It probably is possible to enable GPU compatiability within a conda environment with some effort, but since conda packages binaries, SIMD instructions will not work out of the box.
-
Checkpoints : If you are not sure how long your analysis should run, the standalone script can allow you record checkpoints, which can allow you to recover your model parameters. This enables you to investigate your model while the model is training.
-
More model parameters : The standalone script will return the bias parameters learned for each dataset (i.e. microbe and metabolite abundances). These are stored under the summary directory (specified by
--summary) under the namesembeddings.csv. This file will hold the coordinates for the microbes and metabolites, along with biases. There are 4 columns in this file, namelyfeature_id,axis,embed_typeandvalues.feature_idis the name of the feature, whether it be a microbe name or a metabolite feature id.axiscorresponds to the name of the axis, which either corresponds to a PC axis or bias.embed_typedenotes if the coordinate corresponds to a microbe or metabolite.valuesis the coordinate value for the givenaxis,embed_typeandfeature_id. This can be useful for accessing the raw parameters and building custom biplots / ranks visualizations - this also has the advantage of requiring much less memory to manipulate.
It is also important to note that you don't have to explicitly choose - it is very doable to run the standalone version first, then import those output files into qiime2. Importing can be done as follows
qiime tools import --input-path <your ranks file> --output-path conditionals.qza --type FeatureData[Conditional]
qiime tools import --input-path <your ordination file> --output-path ordination.qza --type 'PCoAResults % Properties("biplot")'
Q : You mentioned that you can use GPUs. How can you do that??
A : This can be done by running pip install tensorflow-gpu in your environment. See details here.
At the moment, these capabilities are only available for the standalone CLI due to complications of installation. See the --arm-the-gpu option in the standalone interface.
Q : Neural networks scare me - don't they overfit the crap out of your data?
A : Here, we are using shallow neural networks (so only two layers). This falls under the same regime as PCA and SVD. But just as you can overfit PCA/SVD, you can also overfit mmvec. Which is why we have Tensorboard enabled for diagnostics. You can visualize the cv_rmse to gauge if there is overfitting -- if your run is strictly decreasing, then that is a sign that you are probably not overfitting. But this is not necessarily indicative that you have reach the optimal -- you want to check to see if logloss has reached a plateau as shown above.
Q : I'm confused, what is Tensorboard?
A : Tensorboard is a diagnostic tool that runs in a web browser - note that this is only explicitly supported in the standalone version of mmvec. To open tensorboard, make sure you’re in the mmvec environment and cd into the folder you are running the script above from. Then run:
tensorboard --logdir .
Returning line will look something like:
TensorBoard 1.9.0 at http://Lisas-MacBook-Pro-2.local:6006 (Press CTRL+C to quit)
Open the website (highlighted in red) in a browser. (Hint; if that doesn’t work try putting only the port number (here it is 6006), adding localhost, localhost:6006). Leave this tab alone. Now any mmvec output directories that you add to the folder that tensorflow is running in will be added to the webpage.
If working properly, it will look something like this
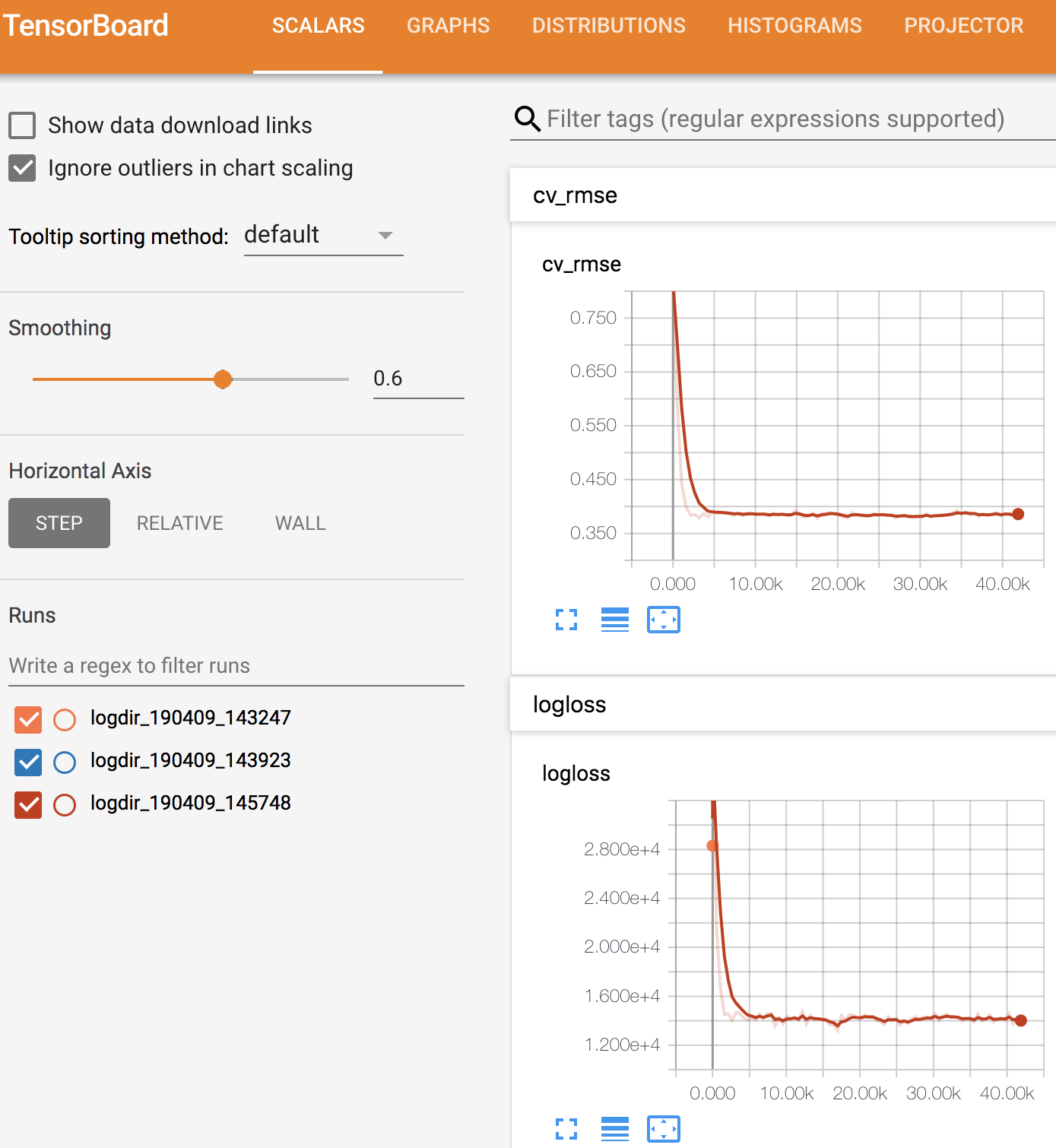
FIRST graph in Tensorflow; 'Prediction accuracy'. Labelled cv_rmse
This is a graph of the prediction accuracy of the model; the model will try to guess the metabolite intensitiy values for the testing samples that were set aside in the script above, using only the microbe counts in the testing samples. Then it looks at the real values and sees how close it was.
The second graph is the likelihood - if your likelihood values are plateaued, that is a sign that you have converged and reached at a local minima.
The x-axis is the number of iterations (meaning times the model is training across the entire dataset). Every time you iterate across the training samples, you also run the test samples and the averaged results are being plotted on the y-axis.
The y-axis is the average number of counts off for each feature. The model is predicting the sequence counts for each feature in the samples that were set aside for testing. So in the graph above it means that, on average, the model is off by ~0.75 intensity units, which is low. However, this is ABSOLUTE error not relative error (unfortunately we don't know how to compute relative errors because of the sparsity in these datasets).
You can also compare multiple runs with different parameters to see which run performed the best. Useful parameters to note are --epochs and --batch-size. If you are committed to fine-tuning parameters, be sure to look at the training-column example make the testing samples consistent across runs.
Q : What's up with the --training-column argument?
A : That is used for cross-validation if you have a specific reproducibility question that you are interested in answering. It can also make it easier to compare cross validation results across runs. If this is specified, only samples labeled "Train" under this column will be used for building the model and samples labeled "Test" will be used for cross validation. In other words the model will attempt to predict the microbe abundances for the "Test" samples. The resulting prediction accuracy is used to evaluate the generalizability of the model in order to determine if the model is overfitting or not. If this argument is not specified, then 10 random samples will be chosen for the test dataset. If you want to specify more random samples to allocate for cross-validation, the num-random-test-examples argument can be specified.
Q : What sort of parameters should I focus on when picking a good model?
A : There are 3 different parameters to focus on, input-prior, output-prior and latent-dim
The --input-prior and --output-prior options specifies the width of the prior distribution of the coefficients, where the --input-prior is typically specific to microbes and the --output-prior is specific to metabolites.
For a prior of 1, this means 99% of entries in the embeddings are within -3 and +3 log fold change. A prior of 0.1 would impose the constraint that 99% of the embeddings are within -0.3 and +0.3 log fold change. The higher --input-prior and --output-prior is, the more parameters can have bigger changes, so you want to keep this relatively small for small experimental studies, particularly if there are less than 20 samples (we have not been able to run MMvec on a study with fewer than 12 samples without overfitting).
If you see overfitting (accuracy and fit increasing over iterations in tensorboard) you may consider reducing the --input-prior and --output-prior in order to reduce the parameter space.
Another parameter worth thinking about is --latent-dim, which controls the number of dimensions used to approximate the conditional probability matrix. This also specifies the dimensions of the microbe/metabolite embeddings that are stored in the biplot file. The more dimensions this has, the more accurate the embeddings can be -- but the higher the chance of overfitting there is. The rule of thumb to follow is in order to fit these models, you need at least 10 times as many samples as there are latent dimensions (this is following a similar rule of thumb for fitting straight lines). So if you have 100 samples, you should definitely not have a latent dimension of more than 10. Furthermore, you can still overfit certain microbes and metabolites. For example, you are fitting a model with those 100 samples and just 1 latent dimension, you can still easily overfit microbes and metabolites that appear in less than 10 samples -- so even fitting models with just 1 latent dimension will require some microbes and metabolites that appear in less than 10 samples to be filtered out.
Q : What does a good model fit look like??
A : Again the numbers vary greatly by dataset. But you want to see the both the logloss and cv_rmse curves decaying, and plateau as close to zero as possible.
Q : Should we filter low abundance microbes and metabolites?
A : A rule of thumb that we recommend is to filter out microbes and metabolites that appear in less than 10 samples. The rationale here is that it isn't practical to fit a line with less than 10 samples. By default we filter out microbes that appear in less than 10 samples; this can be controlled by the --min-feature-count option.
Q : How long should I expect this program to run?
A : Both epochs and batch-size contribute to determining how long the algorithm will run, namely
Number of iterations = epoch # multiplied by the ( Total # of microbial reads / batch-size parameter)
This also depends on if your program will converge. The learning-rate specifies the resolution, smaller step size = smaller resolution, which will increase the accuracy, but may take longer to converge. You may need to consult with Tensorboard to make sure that your model fit is sane. See this paper for more details on gradient descent: https://arxiv.org/abs/1609.04747
If you are running this on a CPU, 16 cores, a run that reaches convergence should take about 1 day.
If you have a GPU - you maybe able to get this down to a few hours. However, some finetuning of the batch-size parameter maybe required -- instead of having a small batch-size < 100, you'll want to bump up the batch-size to between 1000 and 10000 to fully leverage the speedups available on the GPU.
As a good reference, the cystic fibrosis dataset can be processed within 10 minutes on a single CPU and within 1 minute on a GPU.
Q : Can I run the standalone version of mmvec and import those outputs to visualize in qiime2?
A : Yes you can! If you ran the standalone mmvec paired-omics command and you specified your ranks and ordination to be stored under conditionals.tsv and ordination.txt, you can import those as qiime2 Artifacts as follows.
qiime tools import --input-path conditionals.tsv --output-path ranks.qza --type "FeatureData[Conditional]"
qiime tools import --input-path ordination.txt --output-path biplot.qza --type "PCoAResults % Properties('biplot')"
Q : Can MMvec handle small sample studies?
A : We have ran MMvec with published studies as few as 19 samples. However running MMvec in these small sample regimes requires careful tuning of --latent-dimension in addition to the --input-prior and --output-prior commands. The desert biocrust experiment maybe a good dataset to refer to when analyzing these sorts of datasets. It is important to note that we have not been able to run MMvec on fewer than 12 samples.
Credits to Lisa Marotz (@lisa55asil), Yoshiki Vazquez-Baeza (@ElDeveloper), Julia Gauglitz (@jgauglitz) and Nickolas Bokulich (@nbokulich) for their README contributions.
Q You mentioned that MMvec learns co-occurrence probabilities. How can I extract these probabilities?
A MMvec will output a file of co-occurrence probabilities, where the rows are metabolites and columns are microbes. You can extract the co-occurrence probabilities by applying a softmax transform along the columns. In python, this done as follows
import pandas as pd
from skbio.stats.composition import clr_inv as softmax
ranks = pd.read_table('ranks.txt', index_col=0)
probs = ranks.apply(softmax)
probs.to_csv('conditional_probs.txt', sep='\t')
Citation
If you found this tool useful please cite us at
@article{morton2019learning,
title={Learning representations of microbe--metabolite interactions},
author={Morton, James T and Aksenov, Alexander A and Nothias, Louis Felix and Foulds, James R and Quinn, Robert A and Badri, Michelle H and Swenson, Tami L and Van Goethem, Marc W and Northen, Trent R and Vazquez-Baeza, Yoshiki and others},
journal={Nature methods},
volume={16},
number={12},
pages={1306--1314},
year={2019},
publisher={Nature Publishing Group}
}
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file mmvec-1.0.6.tar.gz.
File metadata
- Download URL: mmvec-1.0.6.tar.gz
- Upload date:
- Size: 39.2 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.2.0 pkginfo/1.6.1 requests/2.24.0 setuptools/49.6.0.post20200814 requests-toolbelt/0.9.1 tqdm/4.48.2 CPython/3.7.8
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
58515472f14ebde94655b25ed7e187a76a6333d3a2990fbd405aa1a77df57f24
|
|
| MD5 |
264f513e8ff2fa9207770be21853c83f
|
|
| BLAKE2b-256 |
936d0a9ea08aad48c157e6261e257e3c46337cb3d483b090ae373e2ff13f5752
|







