MPA: MoBiDiC Prioritization Algorithm

Project description
MPA: MoBiDiC Prioritization Algorithm





Overview
The MPA is a prioritizing algorithm for Next Generation Sequencing molecular diagnosis. We propose an open source and free for academic user workflow.
Variant ranking is made with a unique score that take into account curated database, biological assumptions, splicing predictions and the sum of various predictors for missense alterations. Annotations are made for exonic and splicing variants up to +300nt.
We show the pertinence of our clinical diagnosis approach with an updated evaluation of in silico prediction tools using DYSF, DMD, LMNA, NEB and TTN variants from the human expert-feeded Universal Mutation Database [1] with courtesy regards of curators for pathogenic variants and from the ExAc database [2] to define the dataset of neutral variants.
MPA needs an annotated vcf by ANNOVAR and give as output an annotated vcf with MPA score & ranks.
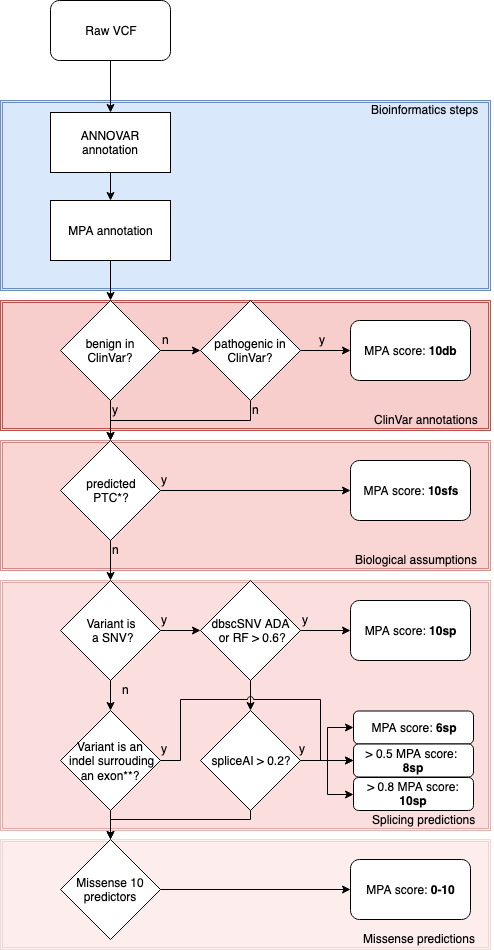
*PTC: Premature Truncation Codon : nonsense or frameshift or start loss
**: intronic positions between -20 and +5
Input
The MPA uses, as input, an annotated VCF file with Annovar [3] and the following databases :
- Curated database: ClinVar [4]
- Biological assumption : refGene [5]
- Splicing predicition : SpliceAI [6], dbscSNV [7]
- Missense prediction : dbNSFP [8]
Note : Short tutorial to annotate your VCF with Annovar (cf. Quick guide for Annovar).
Update April 2019: spliceAI annotations now replace spidex. Waiting for spliceAI to be included in ANNOVAR, Files for this dataset in the proper format are available upon request (hg19 or hg38).
Multi-allelic variants in vcf should be splitted to biallelic variants with bcftools norm.
bcftools norm -m - file.vcf > file_breakmulti.vcf
Output
In a VCF format
VCF is annotated with multiples items : MPA_impact (Clinvar_pathogenicity, splice_impact, stop and frameshift_impact, missense_impact and unknown_impact), MPA_ranking (1 to 8), MPA_final_score (from 0 to 10) and details for the scoring as MPA_available (from 0 to 10 missense tools which annotate), MPA_deleterious (number of missense tools that annotate pathogenic), MPA_adjusted (normalize missense score from 0 to 10).
Ranking : from 1 to 10 and score
- 1. clinvar_pathogenicity : Pathogenic variants reported on ClinVar (score : 10)
- 2. stop or frameshift_impact or start loss: Premature Truncation Codon : nonsense or frameshift (score : 10)
- 3. splicing_impact (ADA, RF) : Affecting splice variants predictions ranked by algorithm performance robustness and strength (score : 10)
- 4. splicing_impact (spliceAI high) : Affecting splice variants predictions ranked by algorithm performance robustness and strength (score : 10)
- 5. missense impact moderate to high impact (6-10)
- 6. moderate splicing_impact (spliceAI moderate) (score 6)
- 7. missense_impact moderate : Missense variants scores low impact (score : 2-6)
- 8. low splicing impact (spliceAI low) or exonic nonframeshift_impact (indel) (score : 2)
- 9. missense_impact low : Missense variants scores low impact (score : 0-2)
- 10. unknown impact : Exonic variants with not clearly annotated ORFs and splicing variants not predicted pathogenic ; or NULL (no annotation on genes, splice etc...) (score : 0-10)
With a simple interface (Captain ACHAB)
MPA is a part of MobiDL captainAchab workflow. MPA is the core of ranking in our useful and simple interface to easily interpret NGS variants at a glance named Captain ACHAB. Find more informations at Captain ACHAB
Installation
Conda



With an activated Bioconda channel (see 2. set up channels), install with:
conda install -c bioconda mobidic-mpa
and update with:
conda update mobidic-mpa
or use the docker container:
docker pull quay.io/biocontainers/mobidic-mpa:<tag>
(see mobidic-mpa/tags for valid values for <tag>)
Pypi




Requirements
- Python >= 3.6
pip
python3 -m pip install mobidic-mpa
Quick start
To run the MPA script, use this command line :
mpa -i path/to/input.vcf -o path/to/output.vcf
Quick guide for Annovar
This algorithm introduce here need some basics annotation. We introduce here a quick guide to annotate your VCF files with Annovar.
Install Annovar
Follow instruction to download Annovar at :
http://www.openbioinformatics.org/annovar/annovar_download_form.php
Unpack the package by using this command :
tar xvfz annovar.latest.tar.gz
Download all databases
In Annovar folder, download all database needed with annotate_variation.pl:
perl annotate_variation.pl -buildver hg19 -downdb -webfrom annovar refGeneWithVer humandb/
perl annotate_variation.pl -buildver hg19 -downdb -webfrom annovar clinvar_20190305 humandb/
perl annotate_variation.pl -buildver hg19 -downdb -webfrom annovar dbnsfp35a humandb/
perl annotate_variation.pl -buildver hg19 -downdb -webfrom annovar dbscsnv11 humandb/
Deprecated: For Spidex database, follow instruction here :
http://www.openbioinformatics.org/annovar/spidex_download_form.php
Update April 2019: spliceAI annotations now replace spidex. Waiting for spliceAI to be included in ANNOVAR, Files for this dataset in the proper format are available upon request (hg19 or hg38).
Annotate a VCF
The following command line annotate a VCF file :
perl path/to/table_annovar.pl path/to/example.vcf humandb/ -buildver hg19 -out path/to/output/name -remove -protocol refGeneWithVer,refGeneWithVer,clinvar_20190305,dbnsfp35a,spliceai_filtered,dbscsnv11 -operation g,g,f,f,f,f -nastring . -vcfinput -otherinfo -arg '-splicing 20','-hgvs',,,,
Citing MPA
Yauy et al. MPA, a free, accessible and efficient pipeline for SNV annotation and prioritization for NGS routine molecular diagnosis. The Journal of Molecular Diagnostics (2018) https://doi.org/10.1016/j.jmoldx.2018.03.009
Montpellier Bioinformatique pour le Diagnostic Clinique (MoBiDiC)
CHU de Montpellier
France

- Béroud, C. et al. UMD (Universal Mutation Database): 2005 update. Hum. Mutat. 26, 184–191 (2005).
- Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
- Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164–e164 (2010).
- Landrum, M. J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868 (2015).
- O’Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–45 (2016).
- Jaganathan et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 176, 535-548 (2019).
- Jian, X., Boerwinkle, E. & Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 42, 13534–13544 (2014).
- Liu, X., Wu, C., Li, C. & Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 37, 235–241 (2016).
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file mobidic-mpa-1.3.0.tar.gz.
File metadata
- Download URL: mobidic-mpa-1.3.0.tar.gz
- Upload date:
- Size: 13.7 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.0 CPython/3.7.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
10a9c1635dac555abdf71e593b75113b3a87b289778ed916fe776f543bef5524
|
|
| MD5 |
bcc46136787a9d88157c5e25c80d9667
|
|
| BLAKE2b-256 |
805b00a29da89b66dc7eaf99d4ba031b4503981085aaf4e40bd373cfa015dced
|
File details
Details for the file mobidic_mpa-1.3.0-py3-none-any.whl.
File metadata
- Download URL: mobidic_mpa-1.3.0-py3-none-any.whl
- Upload date:
- Size: 11.7 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.0 CPython/3.7.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
978b1f7c204f01571115cd69ac3138aaba4649a4fe279201e2c54ffb122486ff
|
|
| MD5 |
ce1c71b4d860de17006c745845e77374
|
|
| BLAKE2b-256 |
cd53491a3dcb71056b69fa80d8e92241f37f23a0647e8251d10d5acc2c52898b
|







