Generate modbed track files for visualization on WashU Epigenome Browser

Project description
modbedtools
Requires Python >= 3.6
A python command line tool to generate modbed files for visualization on the WashU Epigenome Browser.
This tools has 2 modules/subcommands:
- parse MM/ML tag from BAM/CRAM files generated from 3rd generation sequencing platform like Oxford Nanopore and PacBio devices using the pysam package.
- add background canonical base positions given modified bases.
installation
Install through pypi modbedtools project page (version number might change):
$ pip install modbedtools
Collecting modbedtools
Downloading modbedtools-0.1.3-py3-none-any.whl (8.8 kB)
Requirement already satisfied: pysam in /opt/apps/python3/lib/python3.7/site-packages (from modbedtools) (0.19.1)
Installing collected packages: modbedtools
Successfully installed modbedtools-0.1.3
modbed format
chr11 5173273 5195306 read_id score + -110,-266,-1459,-1780,-1840,-1842,-1848,-1865,-1928,-1936,... -396,-1543,-3222,-4195,-4319,-4692,-5352,-5366,-5523,-5838,...
chr11 5174507 5194585 read_id score + 223,605,607,613,630,693,701,936,1761,3369,... 307,544,1280,2017,2859,2994,3116,3249,3790,3935,...
chr11 5174543 5196481 read_id score + 187,271,508,570,576,593,901,1729,2826,3216,... 568,656,664,1985,2961,3083,3703,4115,4286,4882,...
Each row in this bed-based format is a long read, the columns are:
- chromosome
- start position of this read
- end position of this read
- read name or id or something to tag this read
- score (number), this can be used to sort the reads from top to bottom when viewing in Browser, can use 0 if no need to sort
- strand (+ or - for mapping direction)
- methylated/modified base positions, relative to start, a dot
.can be used if there is no modified bases - unmethylated/unmodified/canonical base positions, relative to start, a dot
.can be used if there is no unmodified bases
All positions are 0 based.
8 columns of data need be provided, 4th column can be read identifiers or use chrom:start-end. 5th column is score which is used to sort reads vertically in the view region.
commands
$ modbedtools -h
usage: modbedtools [-h] [--version] {bam2mod,addbg} ...
Python command line tool to generate modbed files for visualization on WashU Epigenome Browser.
optional arguments:
-h, --help show this help message and exit
--version, -v show program's version number and exit
subcommands:
valid subcommands
{bam2mod,addbg} additional help
bam2mod convert bam to modbed
addbg add backgroud bases given modified bases and reference sequence
(files for testing can be found in the test folder in this repository)
bam2mod
convert bam/cram files with MM/ML tags to modbed format.
$ modbedtools bam2mod -h
usage: modbedtools bam2mod [-h] [-g] [-c CUTOFF] [-r REFERENCE] [-o OUTPUT] bamfile
positional arguments:
bamfile bam/cram file with MM/ML tags
optional arguments:
-h, --help show this help message and exit
-g, --cpg output for both C/G bases in CpG, assumes base is C
-c CUTOFF, --cutoff CUTOFF
methylation cutoff, >= cutoff as methylated. default: 0.5
-r REFERENCE, --reference REFERENCE
reference genome file (required for CRAM files, optional for BAM)
-o OUTPUT, --output OUTPUT
output file name, a suffix .modbed will be added. default: output
examples:
modbedtools bam2mod hifi-test.bam -o hifi
modbedtools bam2mod remora-test.bam -o remora
# CRAM file example (requires reference genome)
modbedtools bam2mod sample.cram -r reference.fa -o sample
addbg
For data provided methylated bases, given a reference genome fasta sequence, add the unmethylated bases from genome sequence as background, this assumes all other specified bases from genome are unmethylated/unmodified.
The input file should be in bed format, the last 2 columns save the comma separated relative base positions with modifications (0 based).
example input:
chr11 5193360 5212743 {middle columns can be anything or none} 21,273,296,307,440,461,475,688,689,694,863...
The example data below is adopted from one of the Fiber-seq data from John Stamatoyannopoulos lab.
modbedtools addbg -b A GSM4411218_tracks_m6A_DS75167.dm6.bed.gz dm6.fa.gz -o GSM4411218_tracks_m6A_DS75167
misc scripts
Convert NanoMethPhase example data to modbed format.
python3 ../misc/parse_nanomethphase.py NA19240_chr21_39000000-40000000.bam NA19240_chr21_39000000-40000000_MethylationCalls.tsv
If need support of other methylation callers please submit an issue request.
track formating
Tabix is used to compress and index the modbed files generated in last steps.
example:
bgzip hifi.modbed
tabix -p bed hifi.modbed.gz
Then the .gz and .gz.tbi files can be placed into any web server for hosting and the URL to the .gz file can be used for Visualization in WashU Epigenome Browser.
changelog
- since version 0.2.0, removed
baseoption forbam2mod
visualization
Example modbed files can be used for visualization:
| File | Description | One-click URL for visualization |
|---|---|---|
| HG00621.remora.modbed.gz | Genome wide ONT remora data | link |
| remora-test-chr11.modbed.gz | ONT remora data only on chr11 | link |
| HG00621.hifi.cpg.modbed.gz | Genome wide PacBio Hifi data | link |
| hifi-test-chr11.cpg.modbed.gz | PacBio Hifi data only on chr11 at CpG mode | link |
| hifi-test.modbed-hbg.gz, index file | PacBio Hifi data only on chr11:5162720-5356331, also for testing local track upload |
link |
| GSM4411218_tracks_m6A_DS75167.dm6.modbed.gz | Fruit fly Fiber-seq data | link |
step by step tutorial
In this tutorial, and we will use hifi-test.modbed.gz for the next step by step tutorial.
(Please note this test data only contains methylation signal on chr11)
First we will go to the Browser by navigating your web browser to https://epigenomegateway.wustl.edu/browser/, click hg38 for the genome.
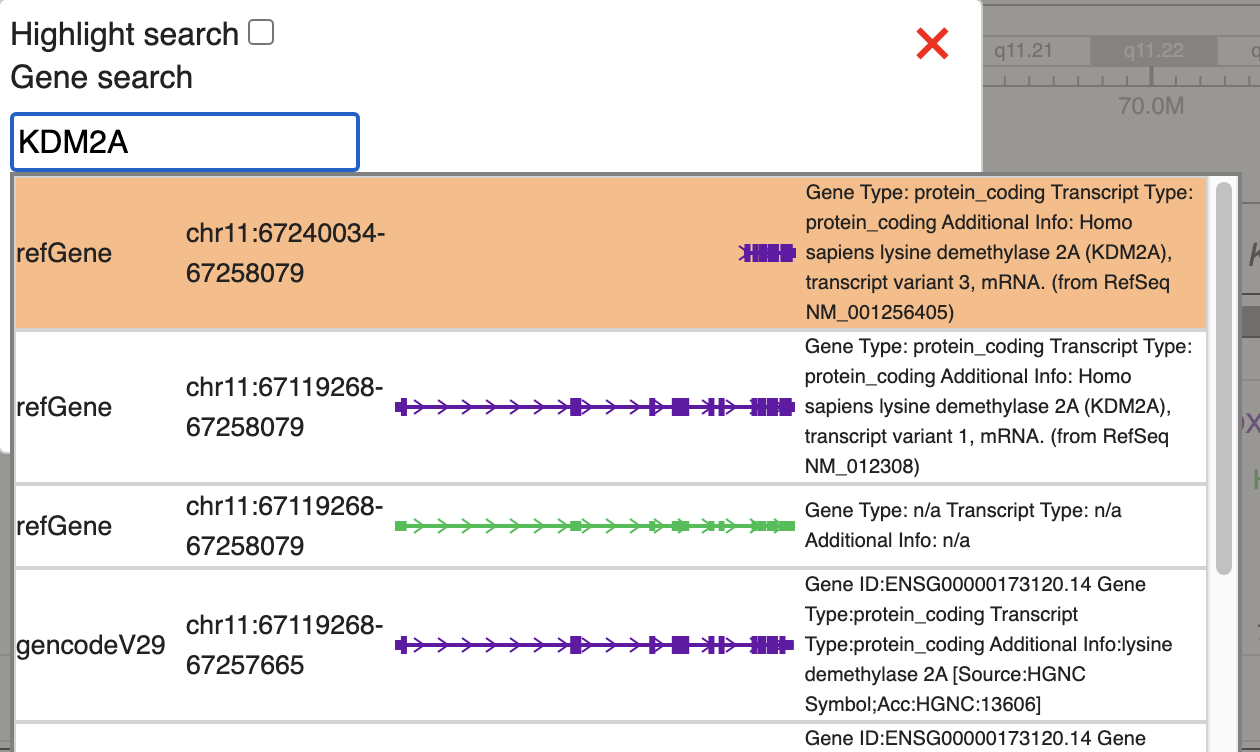
In the test data, we will check methylation signal over KDM2A gene, we will use the gene search function, type in KDM2A and choose the first hit in refGene:
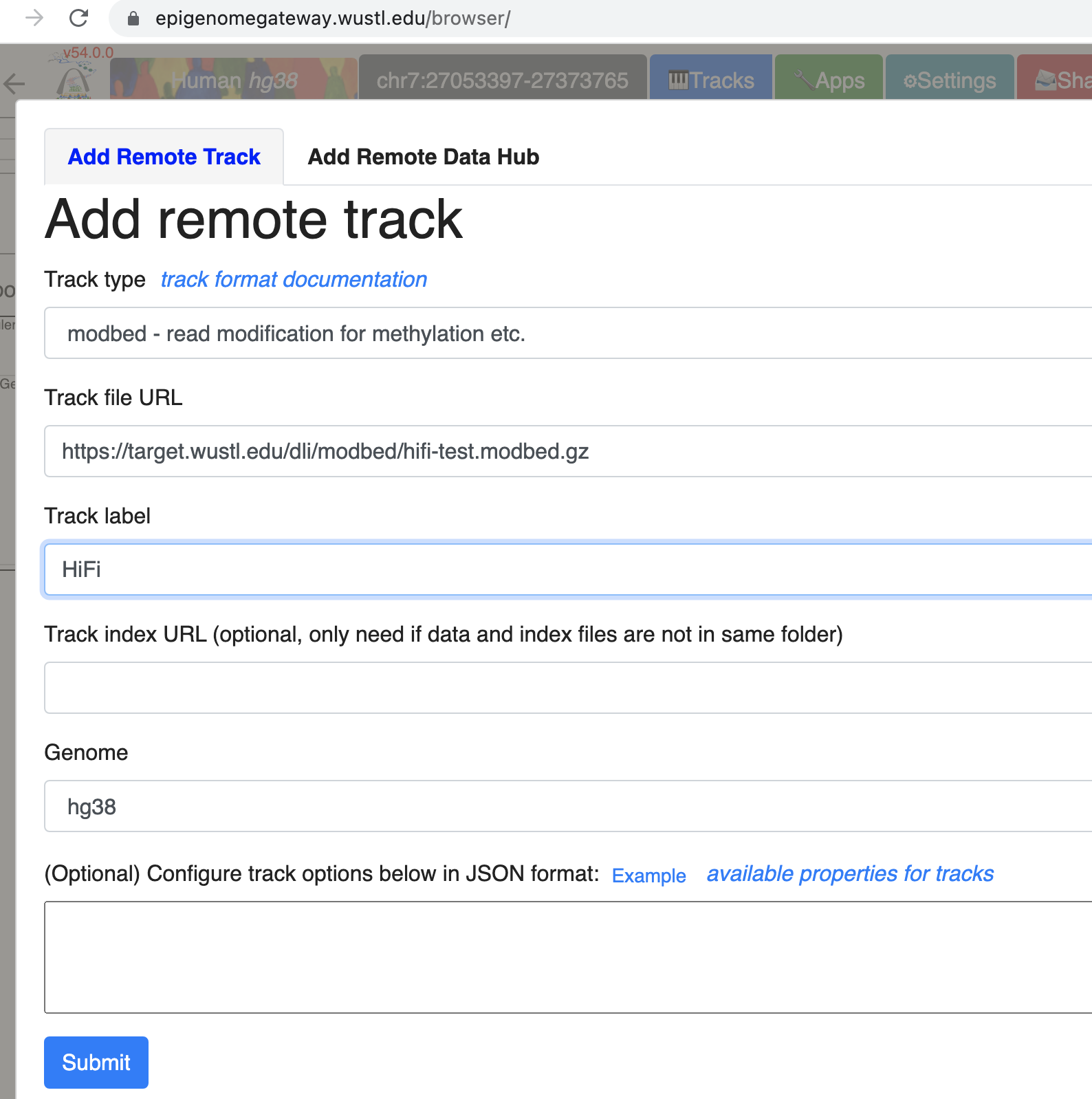
Go to Tracks menu, click Remote Tracks:

Choose modbed from the track type dropdown list, paste the URL above:
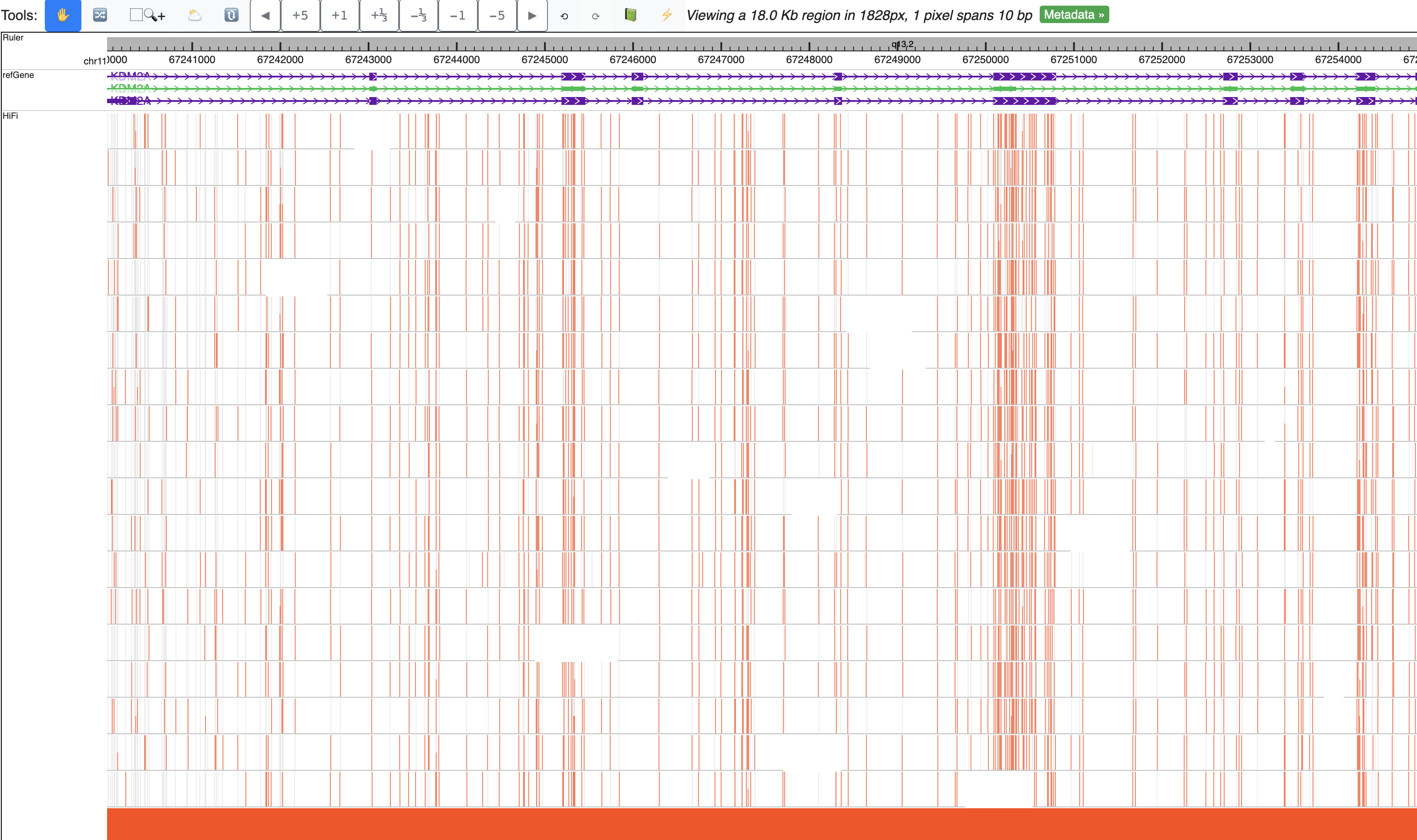
This is the default view after you submit this modbed file, each row represents a long read, each bar on each read means methylation level, gray bar indicates there is an cytosine base but it’s unmethylated. Mouse over each bar can show the tooltip.
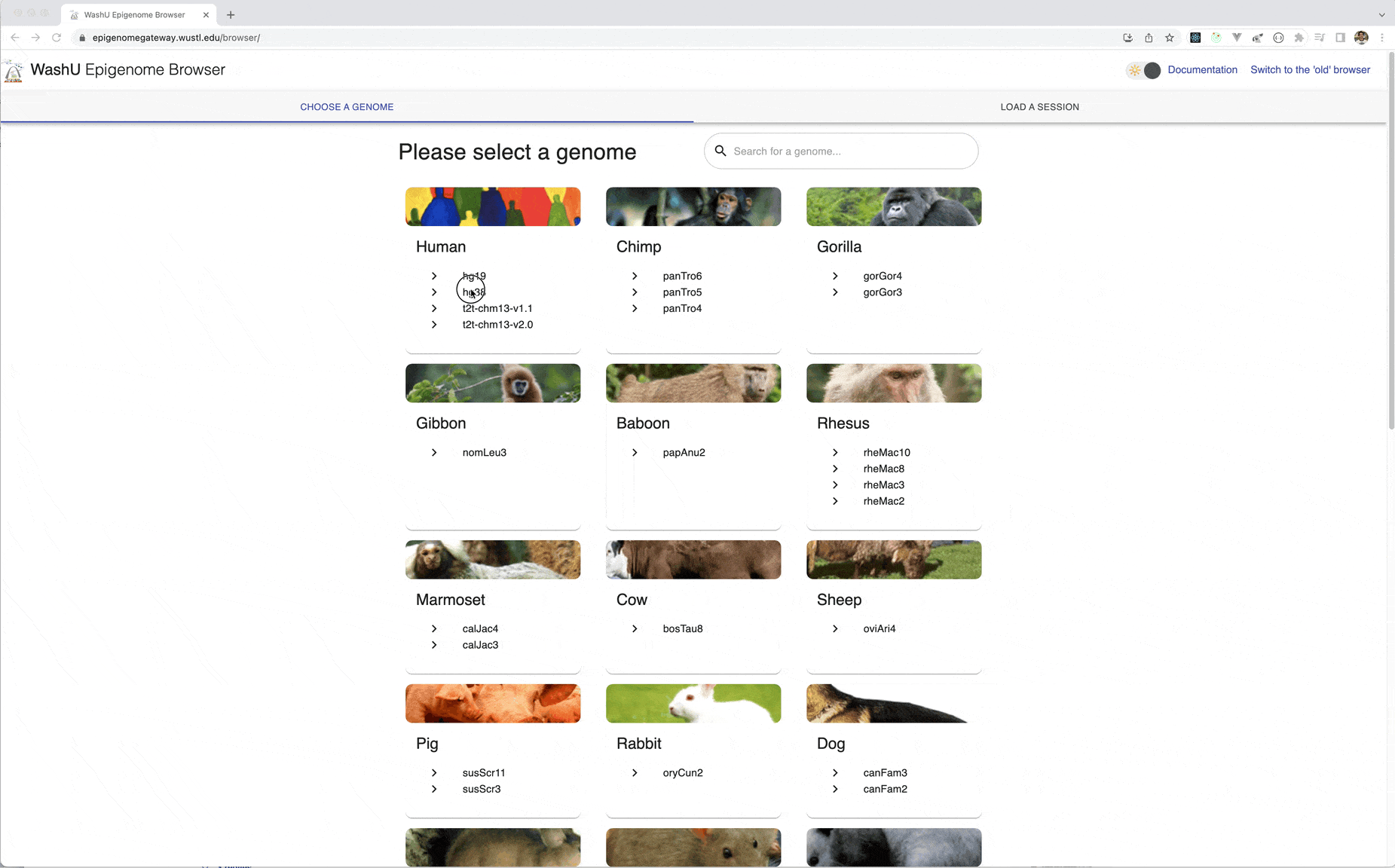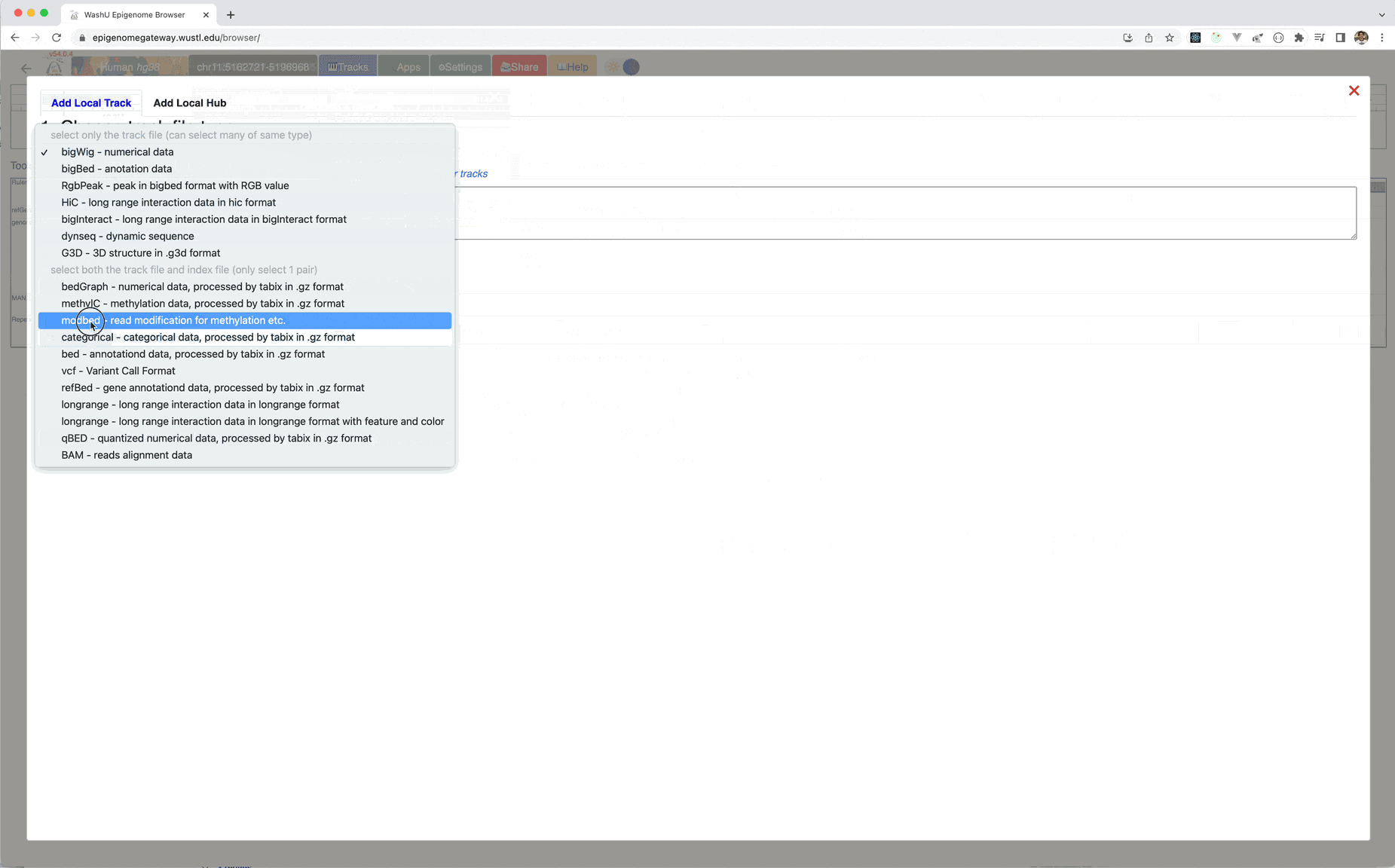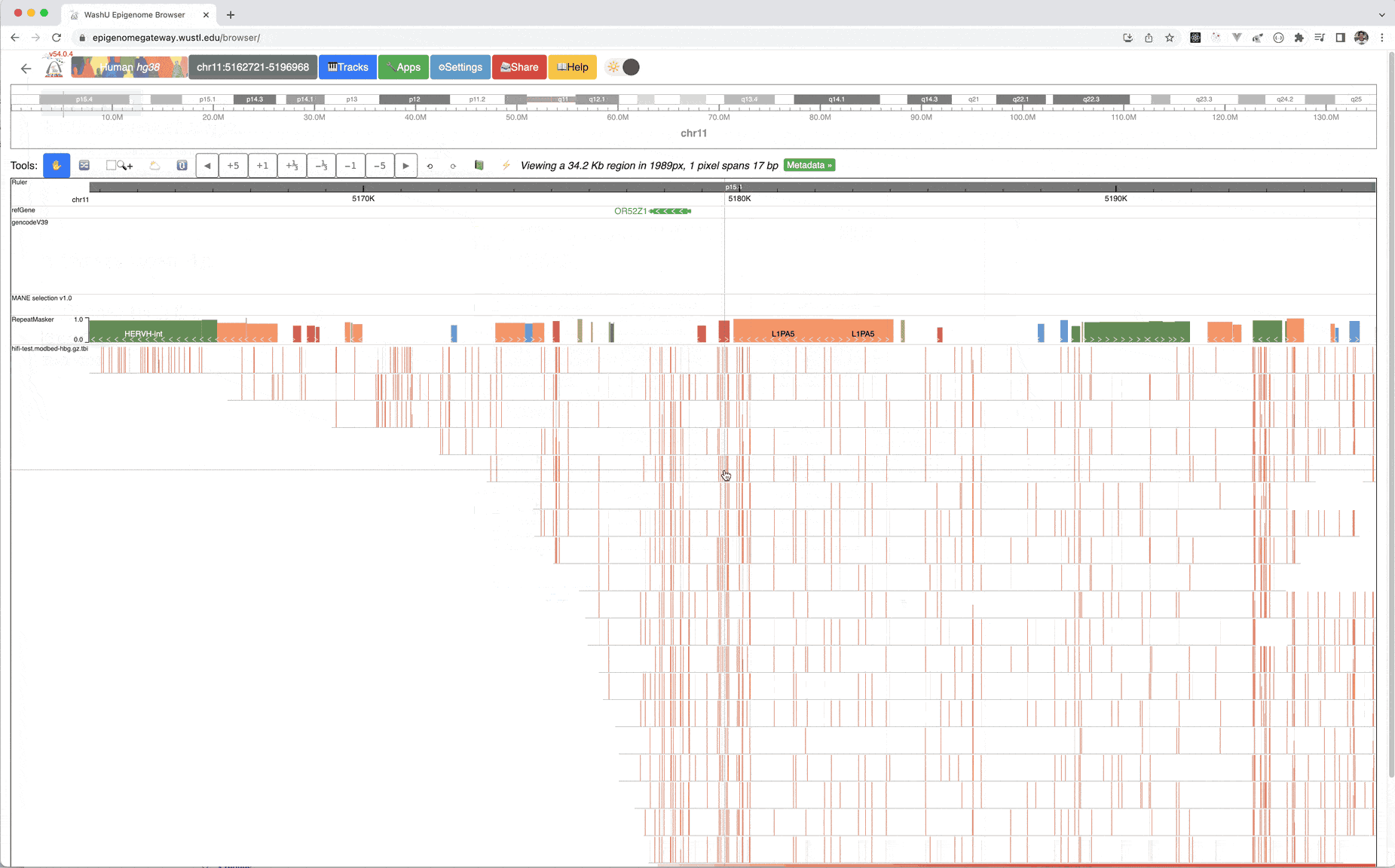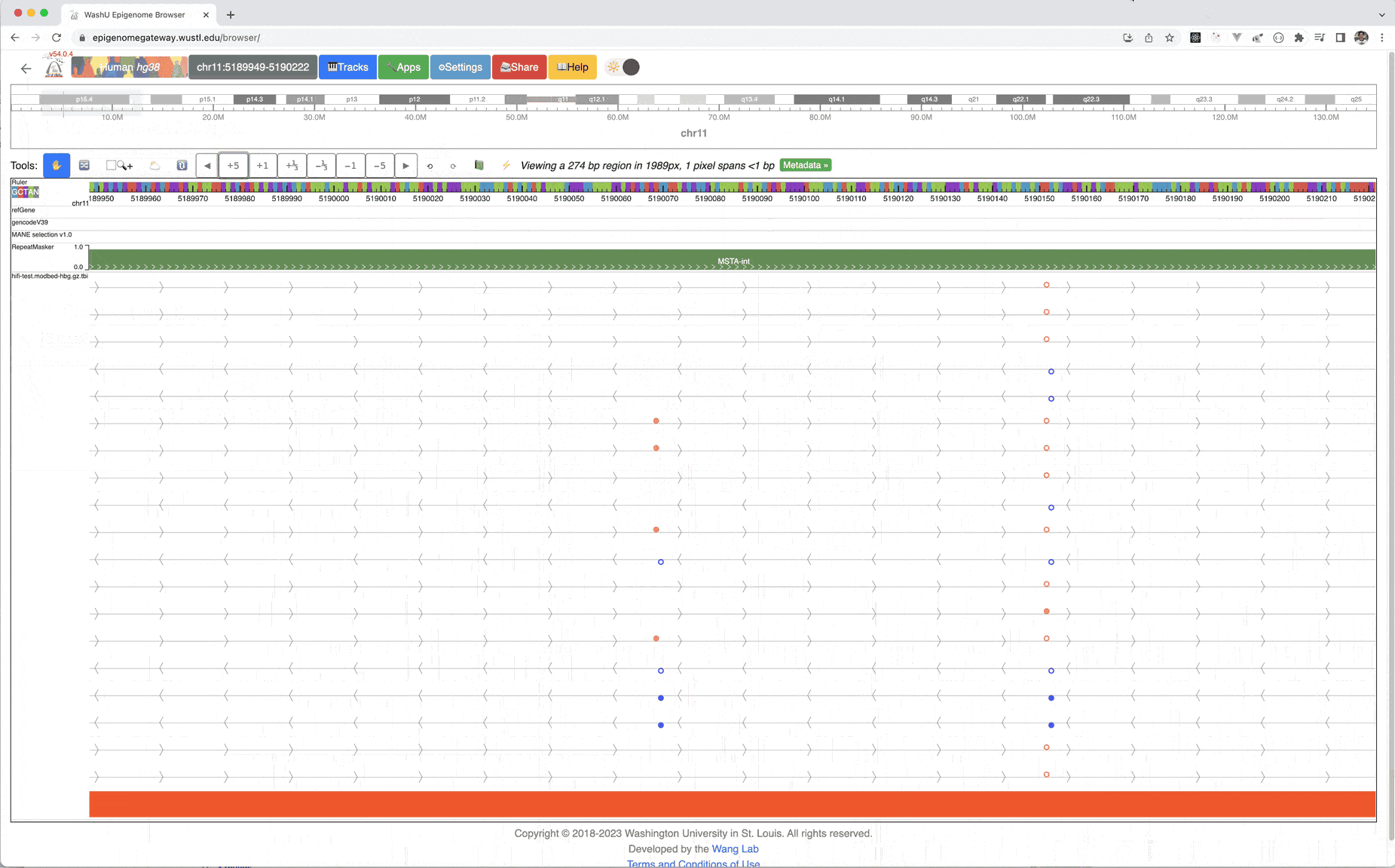
Zoom in 5-fold multiple times, you can see the methylation status at base pair level resolution, each filled circle means methylated, empty circle means unmethylated, orange circle above the line means it’s in + strand, blue in – strand.
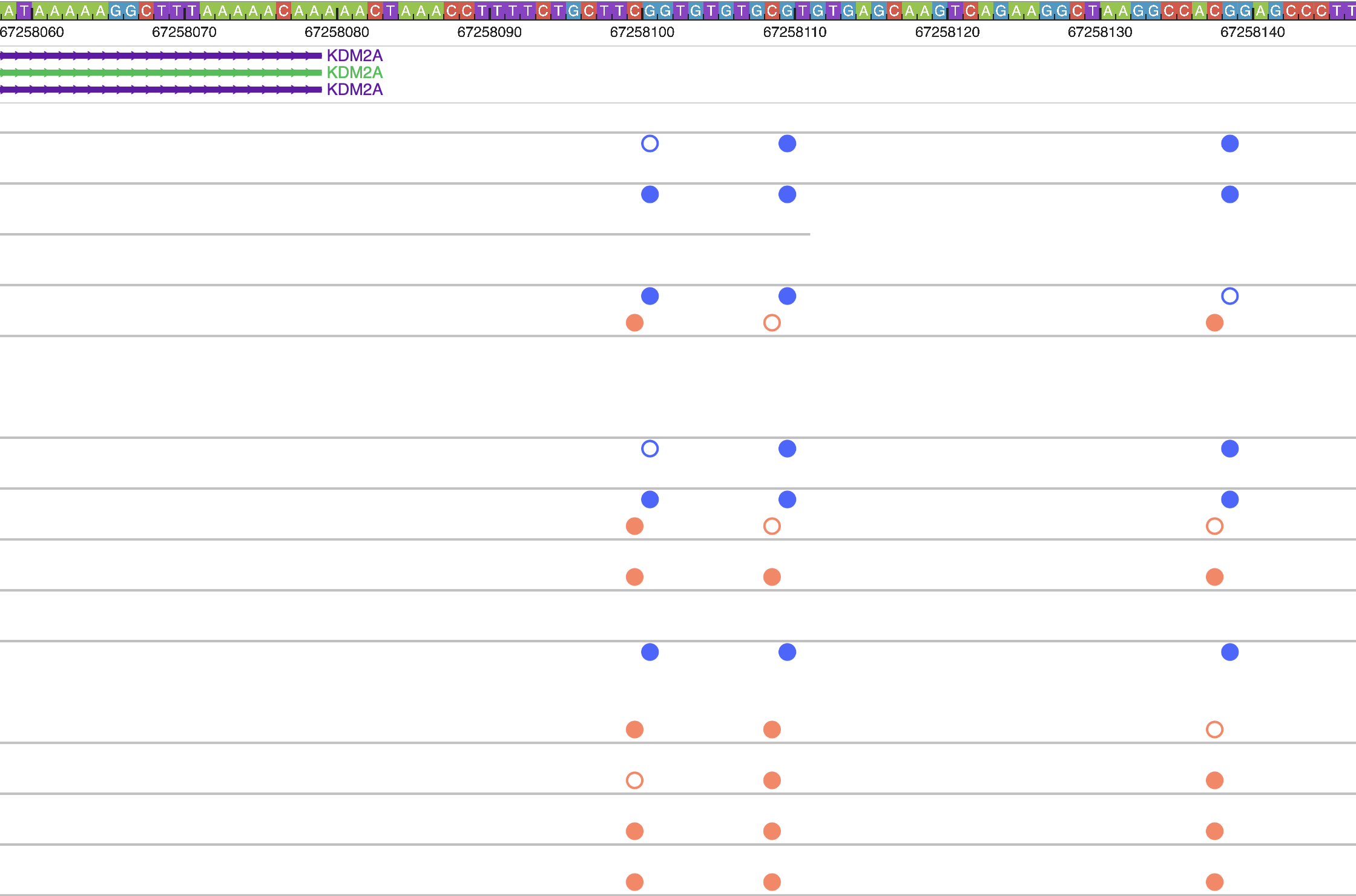
Zoom out multiple times from the default view, can clearly view m6A methylation profile over each read:
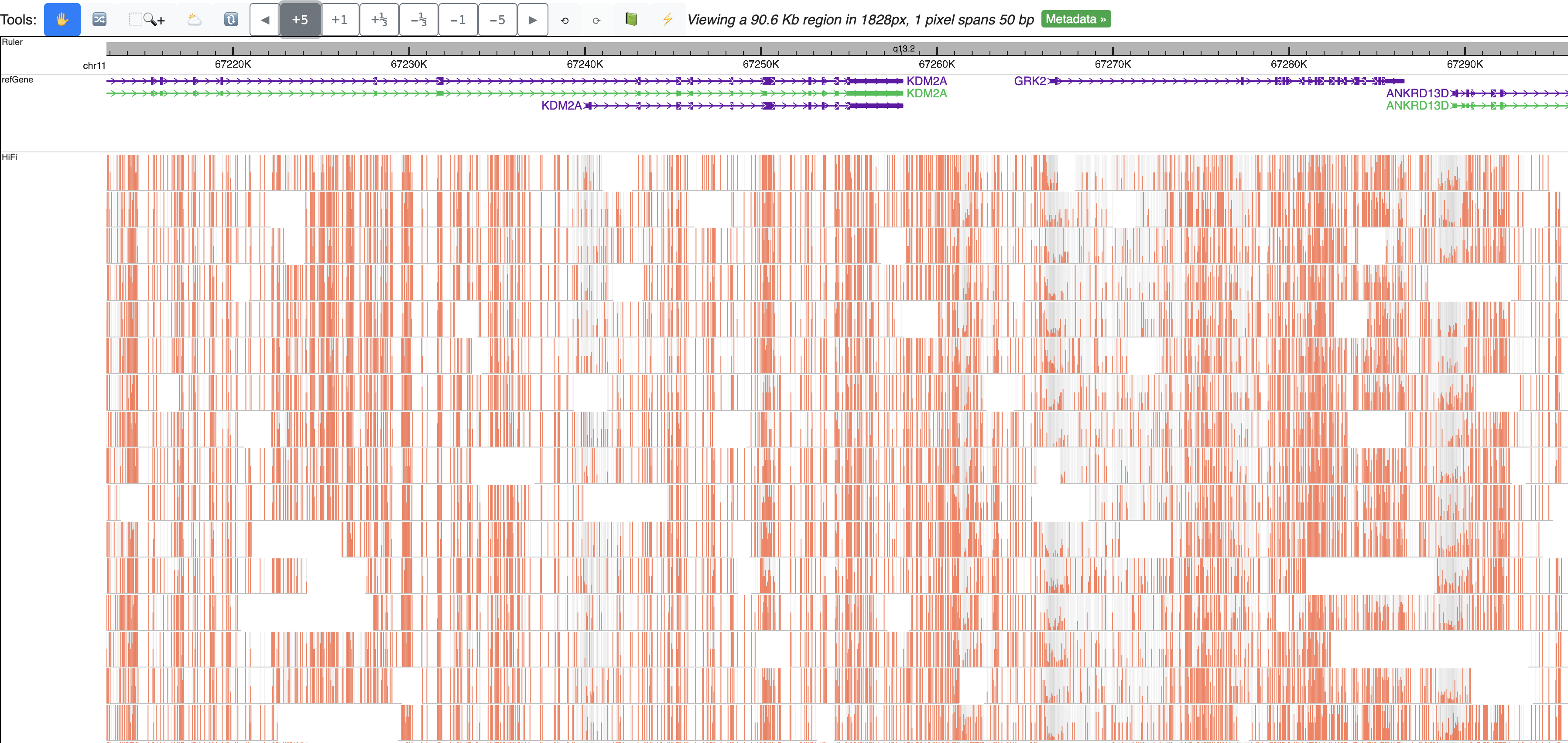
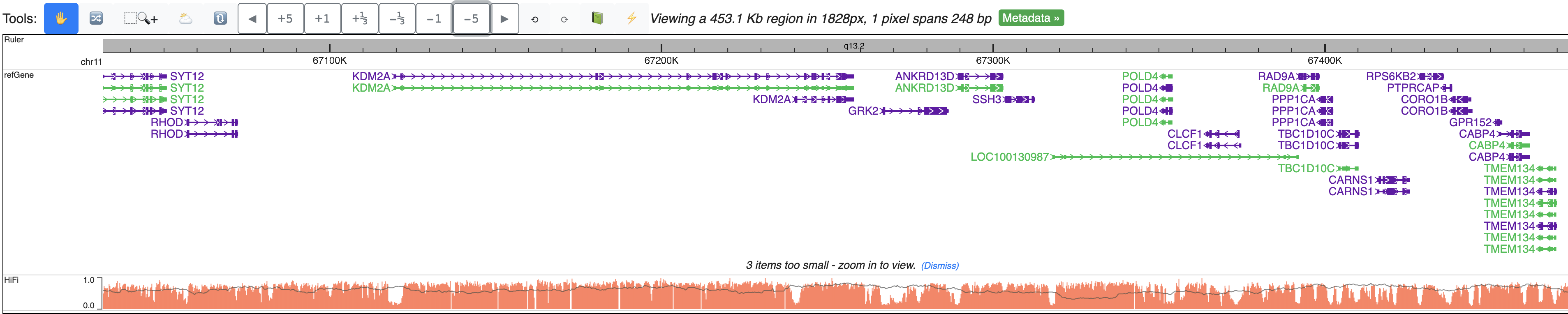
Zoom out further, signals from all reads are summarized to one bar plot, gray line indicates read density, bar height means methylation level:
At each view, right click the track, can change view to heatmap style like in IGV:

upload modbed files as local track
Please see the animation below for instructions, example files can be found here, and the index file, please download both files to your local hard drive.
Pacbio data
PacBio CpG methylation calls of circular consensus se-quencing (ccs) reads represents the predicted methylation status of the CpG site as a unit. Usually, we plotted the methylation prediction of CCS on both C base at each CpG site by enable the -g option:
modbedtools bam2mod hifi-test.bam -o hifi -g
see the below screenshots for pacbio data visualizaed at base pair level, top is without -g and button is with -g option:



Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file modbedtools-0.2.0.tar.gz.
File metadata
- Download URL: modbedtools-0.2.0.tar.gz
- Upload date:
- Size: 13.1 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.11.5
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
1984adafee2371828aba19e84fc61ff3427d9810ec79b60be2aaa104e2d488b2
|
|
| MD5 |
f456ada6c61621e345d7b7d036d7169c
|
|
| BLAKE2b-256 |
3534c4bef7e13ac0f7127223048de74b6944b74578b4bdaf0a984a239596df59
|
File details
Details for the file modbedtools-0.2.0-py3-none-any.whl.
File metadata
- Download URL: modbedtools-0.2.0-py3-none-any.whl
- Upload date:
- Size: 10.2 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.11.5
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
5f1a62cd603e815be4e4ce1e729cb494db544a1119693745644008e3304dbe90
|
|
| MD5 |
f7365d26e636536f8ff3a1561d7b0d95
|
|
| BLAKE2b-256 |
f09fb5aee59b485572efa2a208e3790b368ba08697586b011dcbedafc23cc45d
|







