An all-purpose pythonic genetic algorithm

Project description
NaturalSelection 
An all-purpose pythonic genetic algorithm, which includes built-in hyperparameter tuning support for neural networks.
Installation
$ pip install naturalselection
Usage
Here is a toy example optimising a pair of numbers with respect to division.
>>> import naturalselection as ns
>>>
>>> Pair = ns.Genus(x = range(1, 10000), y = range(1, 10000))
>>>
>>> pairs = ns.Population(
... genus = Pair,
... size = 100,
... fitness_fn = lambda n: n.x/n.y
... )
...
>>> history = pairs.evolve(generations = 100)
Evolving population: 100%|██████████████████| 100/100 [00:05<00:00, 19.59it/s]
>>>
>>> history.fittest
{'genome': {'x': 9922, 'y': 10}, 'fitness': 992.2}
>>>
>>> history.plot()
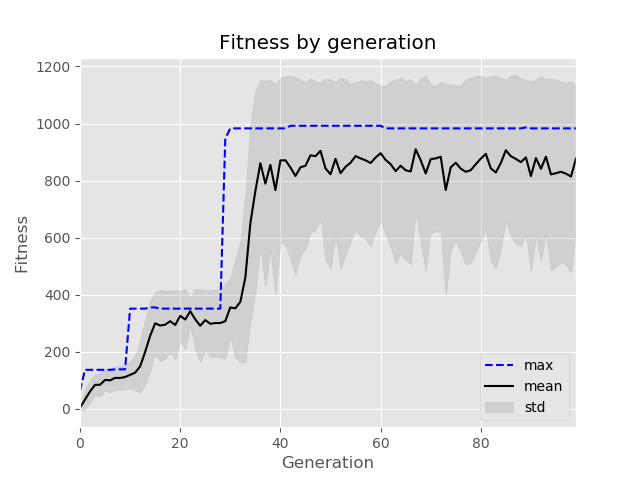
We can also easily solve the classical OneMax problem, which is about finding the bit-string of a given length with all 1's. Here we set goal = 100 in the evolve function to allow for early stopping if we reach our goal before the maximum number of generations, which we here set to 5,000. Note that it only takes nine seconds, however.
>>> import naturalselection as ns
>>>
>>> BitString = ns.Genus(**{f'x{n}' : (0,1) for n in range(100)})
>>>
>>> def sum_bits(bitstring):
... return sum(bitstring.get_genome().values())
...
>>> bitstrings = ns.Population(
... genus = BitString,
... size = 5,
... fitness_fn = sum_bits
... )
...
>>> history = bitstrings.evolve(generations = 500, goal = 100)
Evolving population: 36%|██████ | 1805/5000 [00:09<00:16, 194.43it/s]
>>>
>>> history.plot(only_show_max = True)
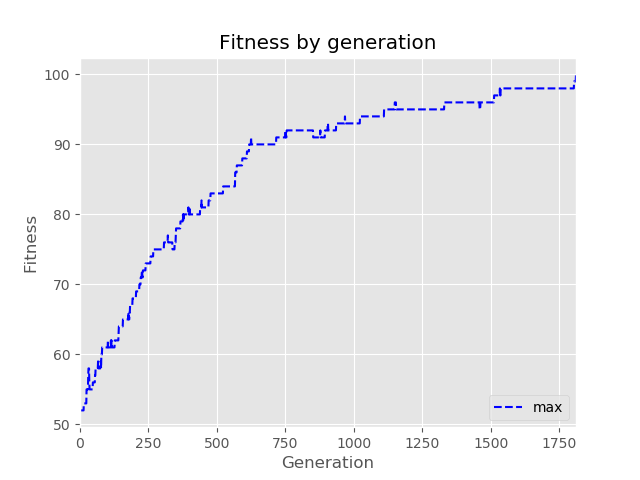
Lastly, here is an example of finding a fully connected feedforward neural network to model MNIST.
Note that the models are trained in parallel, so it is loading in a copy of the MNIST data set for every CPU core in your computer, each of which takes up ~750MB of memory. If this causes you to run into memory trouble then you can set the workers parameter to something small like 2, or set multiprocessing = False to turn parallelism off completely. I've marked these in the code below.
>>> import naturalselection as ns
>>>
>>> def preprocessing(X):
... ''' Basic normalisation and scaling preprocessing. '''
... import numpy as np
... X = X.reshape((-1, np.prod(X.shape[1:])))
... X = (X - X.min()) / (X.max() - X.min())
... X -= X.mean(axis = 0)
... return X
...
>>> def mnist_train_val_sets():
... ''' Get normalised and scaled MNIST train- and val sets. '''
... from tensorflow.keras.utils import to_categorical
... import mnist
... X_train = preprocessing(mnist.train_images())
... Y_train = to_categorical(mnist.train_labels())
... X_val = preprocessing(mnist.test_images())
... Y_val = to_categorical(mnist.test_labels())
... return (X_train, Y_train, X_val, Y_val)
...
>>> nns = ns.NNs(
... size = 30,
... train_val_sets = mnist_train_val_sets(),
... loss_fn = 'categorical_crossentropy',
... score = 'accuracy',
... output_activation = 'softmax',
... max_epochs = 1,
... max_training_time = 60,
... # workers = 2, # If you want to reduce parallelism
... # multiprocessing = False # If you want to disable parallelism
... )
...
>>> history = nns.evolve(generations = 20)
Evolving population: 100%|█████████████████████| 20/20 [57:18<00:00, 73.22s/it]
Computing fitness: 100%|█████████████████████████| 7/7 [01:20<00:00, 10.13s/it]
>>>
>>> history.fittest
{'genome': {'optimizer': 'adagrad', 'hidden_activation': 'relu',
'batch_size': 32, 'initializer': 'glorot_normal', 'input_dropout': 0.2,
'neurons0': 256, 'dropout0': 0.0, 'neurons1': 128, 'dropout1': 0.1,
'neurons2': 256, 'dropout2': 0.1, 'neurons3': 256, 'dropout3': 0.2,
'neurons4': 128, 'dropout4': 0.4}, 'fitness': 0.9659}
>>>
>>> history.plot(
... title = "Validation accuracy by generation",
... ylabel = "Validation accuracy"
... )

We can then train the best performing model and save it locally:
>>> # Training the best model and saving it to mnist_model.h5
>>> best_score = nns.train_best(file_name = 'mnist_model')
Epoch: 0 - loss: 0.273, acc: 0.924, val_loss: 0.116, val_acc: 0.966: 100%|███████| 60000/60000 [00:12<00:00, 1388.45it/s]
(...)
Epoch: 19 - loss: 0.029, acc: 0.991, val_loss: 0.073, val_acc: 0.982: 100%|██████| 60000/60000 [00:11<00:00, 1846.24it/s]
>>>
>>> best_score
0.982
Algorithmic details
The algorithm follows the standard blueprint for a genetic algorithm as e.g. described on this Wikipedia page, which roughly goes like this:
- An initial population is constructed
- Fitness values for all organisms in the population are computed
- A subset of the population (the elite pool) is selected
- A subset of the population (the breeding pool) is selected
- Pairs from the breeding pool are chosen, who will breed to create a new "child" organism with genome a combination of the "parent" organisms. Continue breeding until the children and the elites constitute a population of the same size as the original
- A subset of the children (the mutation pool) is selected
- Every child in the mutation pool is mutated, meaning that they will have their genome altered in some way
- Go back to step 2
We now describe the individual steps in this particular implementation in more detail. Note that step 3 is sometimes left out completely, but since that just corresponds to an empty elite pool I decided to keep it in, for generality.
Step 1: Constructing the initial population
The population is a uniformly random sample of the possible genome values as dictated by the genus, which is run when a new Population object is created. Alternatively, you may set the initial_genome to whatever genome you would like, which will create a population consisting of organisms similar to this genome (the result of starting with a population all equal to the organism and then mutating 80% of them).
>>> pairs = ns.Population(
... genus = Pair,
... size = 100,
... fitness_fn = lambda n: n.x/n.y,
... initial_genome = {'x': 9750, 'y': 15}
... )
...
>>> history = pairs.evolve(generations = 100)
Evolving population: 100%|██████████████████| 100/100 [00:05<00:00, 19.47it/s]
>>>
>>> history.fittest
{'genome' : {'x' : 9989, 'y' : 3}, 'fitness' : 3329.66666666665}
Step 2: Compute fitness values
This happens in the update_fitness function which is called by the evolve function. These computations will by default be computed in parallel when dealing with neural networks and serialised otherwise, as the benefits are only reaped when fitness computations take up a significant part of the algorithm (in the examples above not concerning neural networks we would actually slow down the algorithm non-trivially by introducing parallelism).
Steps 3 & 4: Selecting elite pool and breeding pool
These two pools are selected in exactly the same way, using the sample function. They only differ in the amount of organisms sampled, where the default elitism_rate is 5% and breeding_rate is 80%. In the pool selection it chooses the population based on the distribution with density function the fitness value divided by the sum of all fitness values of the population. This means that the higher fitness score an organism has, the more likely it is for it to be chosen to be a part of the pool. The precise implementation of this is based on the algorithm specified on this Wikipedia page.
Step 5: Breeding
In this implementation the parent organisms are chosen uniformly at random from the breeding pool. When determining the value of the child's genome we apply the "single-point crossover" method, where we choose an index uniformly at random among the attributes, and the child will then inherit all attributes to the left of this index from one parent and the attributes to the right of this index from the other parent.See more on this Wikipedia page.
Step 6: Selection of mutation pool
The mutation pool is chosen uniformly at random in contrast to the other two pools, as otherwise we would suddenly be more likely to "mutate away" many of the good genes of our fittest organisms. The default mutation_rate is 20%.
Step 7: Mutation
This implementation is roughly the bit string mutation, where every gene of the organism has a 1/n chance of being uniformly randomly replaced by another gene, with n being the number of genes in the organism's genome. This means that, on average, mutation causes one gene to be altered. The amount of genes altered in a mutation can be modified by changing thè mutation_factor parameter, which by default is the above 1/n.
Possible future extensions
These are the ideas that I have thought of implementing in the future. Check the ongoing process on the dev branch.
- Enable support for CNNs
- Enable support for RNNs and in particular LSTMs
- Include an option to have dependency relations between genes. In a neural network setting this could include the topology as a gene on which all the layer-specific genes depend upon, which would be similar to the approach taken in this paper.
License
This project is licensed under the MIT License.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file naturalselection-0.6.0.tar.gz.
File metadata
- Download URL: naturalselection-0.6.0.tar.gz
- Upload date:
- Size: 19.9 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/1.13.0 pkginfo/1.5.0.1 requests/2.18.4 setuptools/41.0.1 requests-toolbelt/0.8.0 tqdm/4.19.5 CPython/3.6.8
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
2d2c5dc8251e442c993d255adfb940a0c467f8a45c6434d72514c93aabb42dad
|
|
| MD5 |
609a7d491367714008ca32ea0f36faa3
|
|
| BLAKE2b-256 |
05ba261ac7e768f0e81a5389a2a0997ceee7c7d73f1f3c0bd6e1b081f6d7a338
|
File details
Details for the file naturalselection-0.6.0-py3-none-any.whl.
File metadata
- Download URL: naturalselection-0.6.0-py3-none-any.whl
- Upload date:
- Size: 18.2 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/1.13.0 pkginfo/1.5.0.1 requests/2.18.4 setuptools/41.0.1 requests-toolbelt/0.8.0 tqdm/4.19.5 CPython/3.6.8
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
9e0dee6d6c89a1275f49a4fd8dea70302ac720c89383cbc1680ac418beb83224
|
|
| MD5 |
fe9dbc5fc44efc3987819624fa0c1d38
|
|
| BLAKE2b-256 |
b3ef1af4f1b8c6986170ed75e1326eaa325957871d93731c4ee4a7d81aa554ea
|







