A program to identify transposable element insertions using next generation sequencing data

Project description
ngs_te_mapper2: A program to identify transposable element insertions using next generation sequencing data
Table of Contents
Introduction
ngs_te_mapper2 is a re-implementation of the method for detecting transposable element (TE) insertions from next-generation sequencing (NGS) data originally described in Linheiro and Bergman (2012) PLoS ONE 7(2): e30008. ngs_te_mapper2 uses a three-stage procedure to annotate non-reference TEs as the span of target site duplication (TSD), following the framework described in Bergman (2012) Mob Genet Elements. 2:51-54.
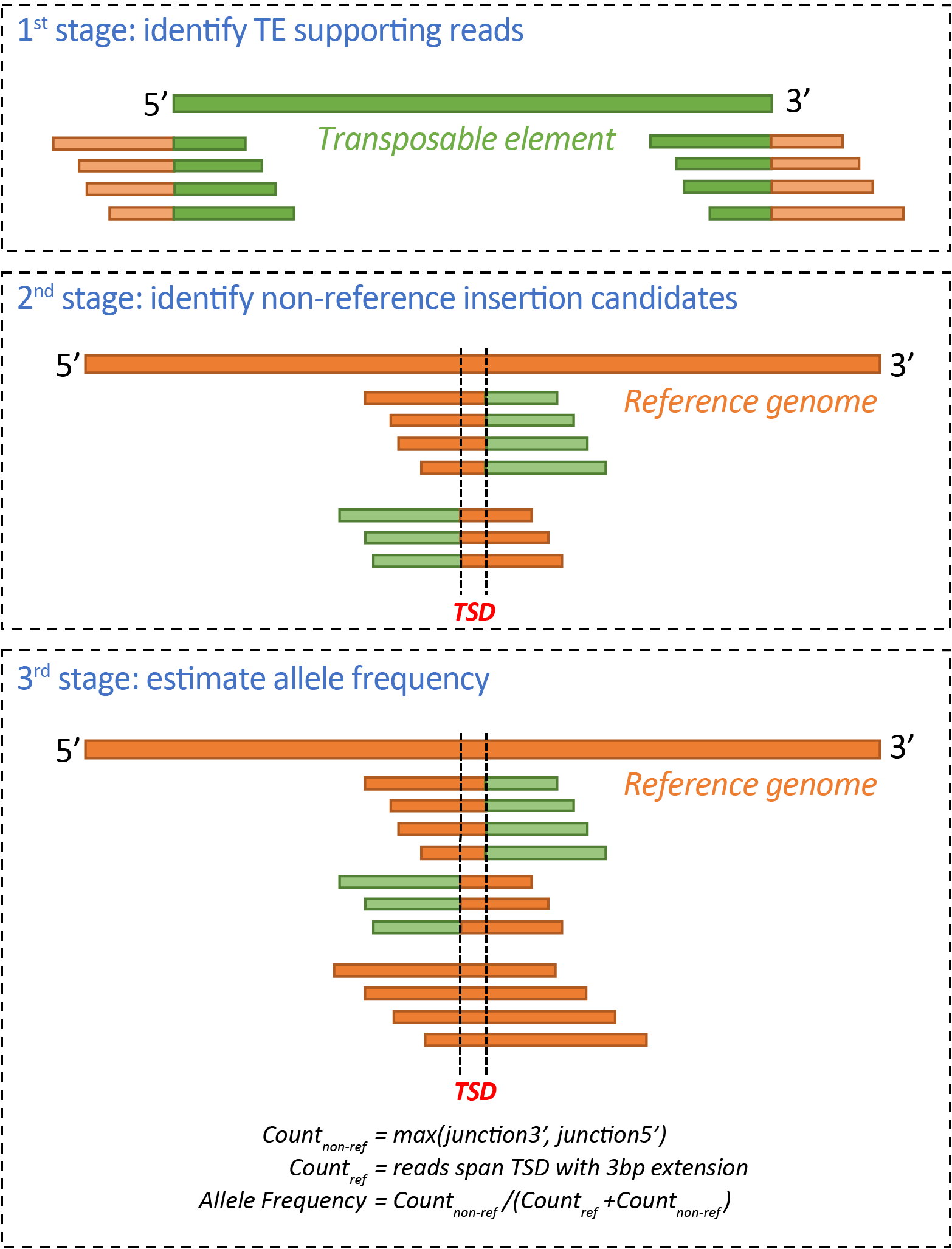
In the first stage, WGS reads are queried against a library of TE sequences to identify 'junction reads' that span the start/end of TE and genomic flanking sequences are retained. Such reads are often referred as 'split reads', although in reality these reads are not split in the resequenced genome.
In the second stage, junction reads from each side of TE insertion identified in the first stage are separately aligned to a reference genome that is hard-masked with RepeatMasker (http://www.repeatmasker.org/) using the same TE library from stage one. Genome-wide coverage profiles are computed using samtools v1.9 and genomic intervals with enriched coverage from junction read clusters on the 5' and 3' side of TEs are annotated in bed format. Regions of overlap between intervals of junction read clusters on the 5' and 3' side of TEs define the locations of the TSDs for candidate non-reference TE insertions. The orientation of the TE is determined from the relative orientation of alignments of the junction reads to the reference genome and TE library.
In the third stage, all reads from the original whole genome shotgun sequence data are used to query against the same hard-masked reference genome as in stage two. This additional mapping step is necessary to obtain all reads that span the TE-flank junction, as well as identify if any reads are present for the alternative ``reference" haplotype that does not carry the TE insertion. For each candidate non-reference TE insertion site, number of junction reads covering 5' and 3' side of each candidate TE insertion are estimated as the number of soft-clipped reads overlapping a 10bp window on the 5' and 3' side of the TSD, respectively (Count_junction5' and Count_junction3'). Number of non-reference reads (Count_non_ref) were estimated as max(Count_junction5', Count_junction3'). Number of reference reads (Count_ref) were estimated as number of non-soft-clipped reads spanning the TSD with at least 3bp extension on both side. The allele frequency for non-reference TEs is heuristically estimated as Count_non-ref/(Count_non_ref + Count_ref).
Reference TE insertions are detected using a similar strategy to non-reference insertions, independently of any reference TE annotation. The first stage in detecting reference TE insertions is identical to the first stage of detecting non-reference TE insertions described above. The second stage in identifying reference TE insertions involves alignment of the renamed, but otherwise unmodified, junction reads to the reference genome. Alignments of the complete junction read (i.e. non-TE and TE components) are clustered to identify the two ends of the reference TE insertion. The orientation of the reference TE is then determined from the relative orientation of alignments of the junction reads to the reference genome and TE library.
Installation
Use Conda to install software dependencies
ngs_te_mapper2 is written in python3 and is designed to run on a Linux operating system. Installation of software dependencies for ngs_te_mapper2 is automated by Conda, thus a working installation of Conda is required to install ngs_te_mapper2. Conda can be installed via the Miniconda installer.
Installing Miniconda (Python 3.X)
wget https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh -O $HOME//miniconda.sh
bash ~/miniconda.sh -b -p $HOME/miniconda # silent mode
echo "export PATH=\$PATH:\$HOME/miniconda/bin" >> $HOME/.bashrc # add to .bashrc
source $HOME/.bashrc
conda init
conda initrequires you to close and open a new terminal before it take effect
Update Conda
conda update conda
Install software dependencies
After installing and updating Conda, you can now use conda to install dependencies and create running environment for ngs_te_mapper2.
Clone ngs_te_mapper2 Repository
git clone git@github.com:bergmanlab/ngs_te_mapper2.git
cd ngs_te_mapper2
Create ngs_te_mapper2 Conda Environment
conda env create -f envs/ngs_te_mapper2.yml
- This installs all the software dependencies needed to run ngs_te_mapper2 into the ngs_te_mapper2 Conda environment.
Activate ngs_te_mapper2 Conda Environment
conda activate ngs_te_mapper2
- This adds the dependencies installed in the ngs_te_mapper2 conda environment to the environment PATH so that they can be used by the ngs_te_mapper2 scripts.
- This environment must always be activated prior to running any of the ngs_te_mapper2 scripts
- NOTE: Sometimes activating conda environments does not work via conda activate myenv when run through a script submitted to a queueing system, this can be fixed by activating the environment in the script as shown below
CONDA_BASE=$(conda info --base)
source ${CONDA_BASE}/etc/profile.d/conda.sh
conda activate ngs_te_mapper2
- For more on Conda: see the Conda User Guide.
Running ngs_te_mapper2 on test dataset
- A test dataset is provided in the
test/directory, you can test whether your ngs_te_mapper2 installation is successful by running ngs_te_mapper2 on this dataset, which should take less than one minute to finish on a single thread machine.
conda activate ngs_te_mapper2
cd test
python3 ../sourceCode/ngs_te_mapper2.py -o test_output -f reads.fastq -r ref_1kb.fasta -l library.fasta
Usage
ngs_te_mapper2 required input files
- FASTQ File (
-f/--reads)- Raw reads from paired-end or single-end sequencing run in fastq or fastq.gz format.
- Multiple fastq/fastq.gz files can be provided separated by comma (ep.
-f R1.fasta,R2.fasta).
- TE library FASTA (
-l/--library)- A FASTA file containing a consensus sequence for each TE family. Note: Each family should only be represented in one sequence in this file.
- Example consensus FASTA file
- Reference FASTA (
-r/--reference)- The genome sequence of the reference genome in FASTA format.
Command line help page
usage: ngs_te_mapper2.py [-h] -f READS -l LIBRARY -r REFERENCE [-a ANNOTATION]
[-n REGION] [-w WINDOW] [--min_mapq MIN_MAPQ]
[--min_af MIN_AF] [--tsd_max TSD_MAX]
[--gap_max GAP_MAX] [-m MAPPER] [-t THREAD] [-o OUT]
[-p PREFIX] [-k]
Script to detect non-reference TEs from single end short read data
required arguments:
-f READS, --reads READS
raw reads in fastq or fastq.gz format, separated by
comma
-l LIBRARY, --library LIBRARY
TE concensus sequence
-r REFERENCE, --reference REFERENCE
reference genome
optional arguments:
-h, --help show this help message and exit
-a ANNOTATION, --annotation ANNOTATION
reference TE annotation in GFF3 format (must have
'Target' attribute in the 9th column)
-w WINDOW, --window WINDOW
merge window for identifying TE clusters (default =
10)
--min_mapq MIN_MAPQ minimum mapping quality of alignment (default = 20)
--min_af MIN_AF minimum allele frequency (default = 0.1)
--tsd_max TSD_MAX maximum TSD size (default = 25)
--gap_max GAP_MAX maximum gap size (default = 5)
-t THREAD, --thread THREAD
thread (default = 1)
-o OUT, --out OUT output dir (default = '.')
-p PREFIX, --prefix PREFIX
output prefix
-k, --keep_files If provided then all intermediate files will be kept
(default: remove intermediate files)
Note: The optional reference TE annotation input should in theory speed up the program. ngs_te_mapper2 expects the TE annotation to be in GFF3 format and Target attribute must be included in the 9th column that represents TE family name. If you have *.out annotation generated by RepeatMasker, you can use this utility script to convert from *.out to GFF3 format.
Output
ngs_te_mapper2 outputs reference and non-referece TE insertion predictions in BED format (0-based).
<sample>.nonref.bed: non-reference TE insertion annotation predicted by ngs_te_mapper2 pipeline in BED format (0-based).<sample>.ref.bed: reference TE insertion annotation predicted by ngs_te_mapper2 pipeline in BED format (0-based).
TE insertion annotation in bed format
ngs_te_mapper2 generates standard BED file <sample>.nonref.bed and <sample>.ref.bed that have detailed information for each reference and non-reference TE insertion.
| Column | Description |
|---|---|
| chromosome | The chromosome name where the TE insertion occurred |
| position | Starting breakpoint position of the TE insertions. |
| end | Ending breakpoint position of the TE insertions. |
| info | Includes TE family, TSD, Allele Frequency, 3' support, 5' support and reference reads. Separated by '|'. |
| score | '.' |
| strand | Strand that TE insertion occurs |
Log file output by ngs_te_mapper2
For each ngs_te_mapper2 run, a log file called <sample>.log is generated that records all the major steps in the program and error messages.
Getting help
Please use the Github Issue page if you have questions.
Citation
To cite ngs_te_mapper2 in publications, please use:
Linheiro, R.S. and C.M. Bergman (2012) Whole Genome Resequencing Reveals Natural Target Site Preferences of Transposable Elements in Drosophila melanogaster. PLoS ONE 7(2): e30008 http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0030008
License
Copyright (c) 2020 Shunhua Han and Casey M. Bergman
Redistribution and use in source and binary forms, with or without modification, are permitted provided that the following conditions are met:
- Redistributions of source code must retain the above copyright notice, this list of conditions and the following disclaimer.
- Redistributions in binary form must reproduce the above copyright notice, this list of conditions and the following disclaimer in the documentation and/or other materials provided with the distribution.
THIS SOFTWARE IS PROVIDED BY THE COPYRIGHT HOLDERS AND CONTRIBUTORS "AS IS" AND ANY EXPRESS OR IMPLIED WARRANTIES, INCLUDING, BUT NOT LIMITED TO, THE IMPLIED WARRANTIES OF MERCHANTABILITY AND FITNESS FOR A PARTICULAR PURPOSE ARE DISCLAIMED. IN NO EVENT SHALL THE COPYRIGHT HOLDER OR CONTRIBUTORS BE LIABLE FOR ANY DIRECT, INDIRECT, INCIDENTAL, SPECIAL, EXEMPLARY, OR CONSEQUENTIAL DAMAGES (INCLUDING, BUT NOT LIMITED TO, PROCUREMENT OF SUBSTITUTE GOODS OR SERVICES; LOSS OF USE, DATA, OR PROFITS; OR BUSINESS INTERRUPTION) HOWEVER CAUSED AND ON ANY THEORY OF LIABILITY, WHETHER IN CONTRACT, STRICT LIABILITY, OR TORT (INCLUDING NEGLIGENCE OR OTHERWISE) ARISING IN ANY WAY OUT OF THE USE OF THIS SOFTWARE, EVEN IF ADVISED OF THE POSSIBILITY OF SUCH DAMAGE.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file ngs_te_mapper2-1.0.2.tar.gz.
File metadata
- Download URL: ngs_te_mapper2-1.0.2.tar.gz
- Upload date:
- Size: 20.5 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.4.2 importlib_metadata/4.4.0 pkginfo/1.7.1 requests/2.26.0 requests-toolbelt/0.9.1 tqdm/4.61.2 CPython/3.6.10
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
825b85095ee76f82e91793769c204253e4ff43cfcd6b3e64ba09784f8d0c2dd3
|
|
| MD5 |
3f306c3bf43f182198ece0b80d077ff5
|
|
| BLAKE2b-256 |
063bd5ce1a73e4451eff759e59de680b783220602353c5ca4c4a39875fe9d59a
|
File details
Details for the file ngs_te_mapper2-1.0.2-py3-none-any.whl.
File metadata
- Download URL: ngs_te_mapper2-1.0.2-py3-none-any.whl
- Upload date:
- Size: 17.8 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.4.2 importlib_metadata/4.4.0 pkginfo/1.7.1 requests/2.26.0 requests-toolbelt/0.9.1 tqdm/4.61.2 CPython/3.6.10
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
a49eebdaa077eda12513d0f2dcf957902d58218daf690def68b6a546f90d61f9
|
|
| MD5 |
66530897a6244b4099b48fd06913e832
|
|
| BLAKE2b-256 |
dc236f92dee8996215859ce0b0513c1311828cc4fd1a76b5f949010892850af7
|







