An exploration of algorithms for phylogeny reconstruction.

Project description
Phylogeny

An exploration on phylogenetic reconstruction algorithms.
In biological evolution one deals with entities that evolve in time. Usually these entities can be represented by strings and the evolution of these entities corresponds to changes in the corresponding string. For example:
An organism may be represented by it’s DNA sequence,
or by a list of characteristics it has
A protein can be represented by it’s nucleotide sequence,
or by it’s sequence of aminoacids.
Although the current package was developed with the above examples in mind, one can think of other similar cases when it would be useful.
The algorithms are based, in part, by the ones found in the books:
Algorithms on Strings, Trees, and Sequences. by Dan Gusfield
Computational Phylogenetics. An introduction to designing methods for phylogeny estimation. by Tandy Warnow
Meta
Author: Ad115 - Github – a.garcia230395@gmail.com
Distributed under the MIT license. See LICENSE for more information.
Contributing
Check for open issues or open a fresh issue to start a discussion around a feature idea or a bug.
Fork the repository on GitHub to start making your changes to a feature branch, derived from the master branch.
Write a test which shows that the bug was fixed or that the feature works as expected.
Send a pull request and bug the maintainer until it gets merged and published.
Tutorial
Instalation
You can install the package from PyPI:
$ pip install phylogeny
Trees, sequences and distance matrices
The main objects we consider are sequences, trees and matrices.
As mentioned before, the entities, represented as sequences, evolve forming trees. At every point of the evolutive process, we can have define a ‘distance matrix’, whose entries are the distance between sequences (a measure of their dissimilarity).
In the phylogeny reconstruction problem, we generally start with a set of sequences that evolved from a common ancestor, or their pairwise distance matrix, and the task is to infer, with the help of certain model assumptions, the tree that generated the evolutionary process.
The Cavender-Farris-Neyman model
Given the nature of the reconstruction problem, an evolution model is the guiding hand behind the inference process. The resulting tree changes depending on the assumptions of the model.
A good model to study reconstruction algorithms is the Cavender-Farris-Neymann (CFN) model, that consists of a random tree with probabilities p(e) associated to every edge e of the tree. Under the CFN model, every character of the sequence or ‘trait’ evolves down the tree changing state on every edge according to the edge probability.
The library contains an implementation of the CFN stochastic tree model for generating sequences from an ancestor.
>>> from phylogeny.models import CFN_Tree
# Create a new random tree with 5 leaves.
>>> cfn = CFN_Tree(leaves=5)
>>> print(cfn)
Node: 0, substitution probability: 0
Node: 1, substitution probability: 0.35099470913056274
Node: 2, substitution probability: 0.12569731001404477
Node: 3, substitution probability: 0.4370228186219944
Node: 4, substitution probability: 0.36030277887942846
Node: 5, substitution probability: 0.4905304532735053
Node: 6, substitution probability: 0.04564697536250617
Node: 7, substitution probability: 0.4178421652970328
Node: 8, substitution probability: 0.2728144976941049
/-3
/1|
| \-4
-0|
| /-7
| /5|
\2| \-8
|
\-6
# Evolve 5 traits through the tree
>>> sequences = cfn.evolve_traits([1,1,1,1,1])
>>> print(sequences)
{3: [0, 0, 1, 1, 0], 4: [1, 1, 1, 1, 1], 7: [1, 1, 1, 1, 1], 8: [1, 0, 0, 1, 1], 6: [1, 1, 1, 1, 1]}
>>> cfn.distance_matrix()
DistanceMatrix([[0. , 1.7, 4.7, 4.2, 1.8],
[1.7, 0. , 4.3, 3.8, 1.4],
[4.7, 4.3, 0. , 1.3, 2.9],
[4.2, 3.8, 1.3, 0. , 2.4],
[1.8, 1.4, 2.9, 2.4, 0. ]], names=(3, 4, 7, 8, 6))The clocklike evolution scenario
The most simple case of evolution is the one in which the evolution is clocklike, that is, that all branches have the same length (the mutation rate is constant over time). In this case, at a certain time, the tree generated has the property that the distance from the root to each leaf is the same.
If one has an ultrametric distance matrix (which represents clocklike evolution), then there are several algorithms to handle the reconstruction, 2 of which are implemented in the library:
>>> from phylogeny import DistanceMatrix
# An ultrametric matrix
>>> ultrametric = DistanceMatrix(
[[0, 8, 8, 5, 3],
[8, 0, 3, 8, 8],
[8, 3, 0, 8, 8],
[5, 8, 8, 0, 5],
[3, 8, 8, 5, 0] ],
names=['A', 'B', 'C', 'D', 'E']
)>>> from phylogeny.reconstruction import infer_clocklike_tree1
>>> t = infer_clocklike_tree1(ultrametric)
>>> print(t)
/-B
/-|
| \-C
--|
| /-A
| /-|
\-| \-E
|
\-D>>> from phylogeny.reconstruction import infer_clocklike_tree2
>>> t = infer_clocklike_tree2(ultrametric)
>>> print(t)
/-D
/-|
| | /-A
| \-|
--| \-E
|
| /-B
\-|
\-CClocklike reconstruction on the CFN model
Now we can try to apply the clocklike assumption to a CFN model.
# Create a new random tree with 5 leaves.
>>> cfn = CFN_Tree(leaves=5)
>>> cfn.show()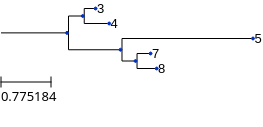
# Evolve traits through the tree
>>> sequences = cfn.evolve_traits([1]*10_000)
# Get the distance matrix
>>> distances = DistanceMatrix.from_sequences(sequences)
# Infer the tree
>>> t = infer_clocklike_tree1(distances)
>>> print(t)
/-8
--|
| /-5
\-|
| /-7
\-|
| /-3
\-|
\-4We can see that the different branch lengths from the root to each leaf confuses the algorithm and we get a tree that is not correct. (The reconstruction thus depends mostly on branch length, not on the topology of the original tree)
Clocklike reconstruction for biological evolution
Now we test the hypothesis on a simulation of biological microevolution.
# Download from PyPI:
# pip install cellsystem
>>> from cellsystem import *
# The cell system will simulate cell growth
# while tracking the steps in that process.
>>> system = CellSystem(init_genome='A'*70)
# Initialize the first cell
# in the middle of the grid
>>> system.seed()
# Take 20 steps forward in time
>>> system.run(steps=5)
# Stop logging the steps to the screen
>>> system.log['printer'].silence()
>>> system.run(steps=15)New cell 0 added @ (50, 50)
Cell no. 0 migrating from site (50, 50) (father None)
New site: (51, 49)
Cell no. 0 migrating from site (51, 49) (father None)
New site: (51, 48)
Cell no. 0 dividing @ (51, 48)
New cells: 1 @ (52, 48) and 2 @ (51, 48)
Cell no. 1 mutating @ site (52, 48) (father None)
Initial mutations: []
Initial genome: AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA
Final mutations: [(65, 'G')]
Final genome: AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGAAAA
# Look at the real ancestry tree
>>> t = system.log.ancestry(prune_death=True)
>>> print(t)
/-9
/-|
| \-10
--|
| /-7
\-|
\-8
# Fetch the evolved DNA sequences
>>> cell_sequences = {cell.index:cell.genome for cell in system['cells'].alive_cells}
# Get the distance matrix
>>> distances = DistanceMatrix.from_sequences(cell_sequences)
# Inferr a tree under the clocklike assumption
>>> t = infer_clocklike_tree1(distances)
>>> print(t)
/-10
--|
| /-9
\-|
| /-7
\-|
\-8We can see it works better for this data, although it is not quite there.
The reconstruction problem when evolution is not clocklike
The reconstruction problem when evolution is not clocklike is so hard that one can not even be sure of where the root of the tree goes!! So, in the following, the trees will be fundamentally unrooted, that is, if two trees differ only in the placement of the root, then we can say they are equal.
The four point condition
It must be noted that for every tree there is a distance matrix but not any matrix correspond to a tree, the matrices that do are called ‘additive’. A way to check if a matrix is additive is by checking the Four Point Condition.
To explain the four point condition let’s say we have the following unrooted tree:
1 -\ /- 3
>--<
2 -/ \- 4
Let the distance between leaves a and b be D(a, b). Consider the three following pairwise sums:
D(1, 2) + D(3, 4)
D(1, 3) + D(2, 4)
D(1, 4) + D(2, 3)
The smallest of these sums has to be D(1, 2) + D(3, 4), since it covers all the edges of the tree connecting the four leaves, EXCEPT for the ones on the path separating 1 and 2 from 3 and 4. Furthermore, the two larger of the three pairwise sums have to be identical, since they cover the same set of edges.
The Four Point Condition is the statement that the two largest values of the three pairwise distance sums are the same.
The library contains a check for additivity based on the four point condition:
# Let's take first a matrix that *is* additive
# -- We take the matrix representation of a known tree.
>>> distances = cfn.distance_matrix()
>>> distances
DistanceMatrix([[0. , 0.5, 3.1, 1.5, 1.6],
[0.5, 0. , 3.4, 1.7, 1.8],
[3.1, 3.4, 0. , 2.4, 2.5],
[1.5, 1.7, 2.4, 0. , 0.5],
[1.6, 1.8, 2.5, 0.5, 0. ]], names=(3, 4, 5, 7, 8))
>>> print("Distance matrix is additive: ", distances.is_additive())
>>> distances[2,3] += 1
>>> print("Altered matrix is additive: ", distances.is_additive())
Distance matrix is additive: True
Altered matrix is additive: FalseThe four point method
The four point method is based on the four point condition to reconstruct a tree from a 4x4 distance matrix. We calculate the three pairwise sums from the four point condition, we determine which of the three pairwise sums is the smallest, and use that one to define the split for the four leaves into two sets of two leaves each (remember that if D(1,2)+D(3,4) is the smallest sum, then the induced tree must be, in Newick notation, ((1,2),(3,4)).)
# A test matrix to test the four-point method
# L1 -\ /- L2
# >--<
# L3 -/ \- L4
>>> additive = DistanceMatrix([[0, 3, 6, 7],
[3, 0, 7, 6],
[6, 7, 0, 11],
[7, 6, 11, 0]], names=['L1', 'L2', 'L3', 'L4'])>>> from phylogeny.reconstruction import four_point_method
>>> tree = four_point_method(additive, names=additive.names)
>>> print(f"The associated tree is: {tree}")
The associated tree is:
/-L1
/-|
| \-L3
--|
| /-L2
\-|
\-L4The all quartets method
The all quartets method results from the repeated application of the four points method and is useful to reconstruct larger trees.
Given an n×n additive matrix M with n ≥ 5 associated to a binary tree T with positive branch lengths, we can construct T using a two-step technique that we now describe.
In Step 1, we compute a quartet tree on every four leaves by applying the Four Point Method to each 4×4 submatrix of M.
In Step 2, we assemble the quartet trees into a tree on the full set of leaves. Step 1 is straightforward. The technique we use in Step 2 is called the All Quartets Method.
# We start with a known tree
>>> cfn = CFN_Tree(leaves=5)
>>> print(cfn)
Node: 0, substitution probability: 0
Node: 1, substitution probability: 0.4465340963982276
Node: 2, substitution probability: 0.1674607139638471
Node: 3, substitution probability: 0.17137831979024587
Node: 4, substitution probability: 0.3491805016323804
Node: 5, substitution probability: 0.12428938378084825
Node: 6, substitution probability: 0.1830304764268481
Node: 7, substitution probability: 0.3684792177646947
Node: 8, substitution probability: 0.3426407225165353
/-1
-0|
| /-3
\2|
| /-5
\4|
| /-7
\6|
\-8from phylogeny.reconstruction import all_quartets_method
# Now we infer it from it's distance matrix
# using the all quartets method
>>> t = all_quartets_method(cfn.distance_matrix())
>>> print(t)
/-5
/-|
| | /-1
| \-|
--| \-3
|
| /-7
\-|
\-8Looks good! Now, let’s test how it performs with the simulated biological sequences
>>> cell_sequences
{7: 'AAAAAAAAAAGAAAAAAAATAAAAAAAAATATAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA',
8: 'AAAAAAAAAAGAAAAAAAATAAAAAAAAATATAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA',
9: 'AAAAAAAAAAGAAAAAAAATAAAAAAAAATATAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA',
10: 'AAAAAAAAAAGAAAAAAAATAAAAAAAAATATAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA'}
>>> m = DistanceMatrix.from_sequences(cell_sequences)
>>> t = all_quartets_method(m)
>>> print(t)
/-7
/-|
| \-8
--|
| /-9
\-|
\-10# Compare with the real ancestry tree
>>> t = system.log.ancestry(prune_death=True)
>>> print(t)
/-9
/-|
| \-10
--|
| /-7
\-|
\-8Looks good too!! If you liked it, please contribute by adding more models, more algorithms, or improving the existing codebase!
If you want to learn more about the algorithms, check the source files or in the reference books.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distributions
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file phylogeny-0.1.1-py3-none-any.whl.
File metadata
- Download URL: phylogeny-0.1.1-py3-none-any.whl
- Upload date:
- Size: 30.5 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/1.9.1 pkginfo/1.4.1 requests/2.18.4 setuptools/39.0.1 requests-toolbelt/0.8.0 tqdm/4.19.6 CPython/3.6.1
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
4769678e96b4aaa10ed8d2ce93f49ddd4374ef5b3ff4b3708d19124b9e3337b5
|
|
| MD5 |
71b34cb3cc4b674a42ac4fc4202e3949
|
|
| BLAKE2b-256 |
78b14b4d2ba07d449e919c07e6fd983980eb928c36e4b4e8e39660bff9d10351
|







