Tree-based orthology inference

Project description

About
Orthologs are genes that are related through a speciation event, while paralogs are genes that are related through a gene duplication event. Accurate identification of orthologs is a prerequisite for phylogenomics, since including genes that diverged because of a gene duplication event for species tree inference can cause an erroneous inference of speciation nodes, due to disparencies between individual gene trees and the species tree. Unfortunately, contaminants present in even a single taxon can cause a tree-based orthology inference method to erroneuosly infer paralogy and unnecessarily exclude sequences.
PhyloPyPruner is a Python package for phylogenetic tree-based orthology inference, using the species overlap method. It uses trees and alignments inferred from the output of a graph-based orthology inference approach, such as OrthoMCL, OrthoFinder or HaMStR, in order to obtain sets of sequences that are 1:1 orthologous. In addition to algorithms seen in pre-existing tree-based tools (for example, PhyloTreePruner, UPhO, Agalma or Phylogenomic Dataset Reconstruction), this package provides new methods for reducing potential contamination.
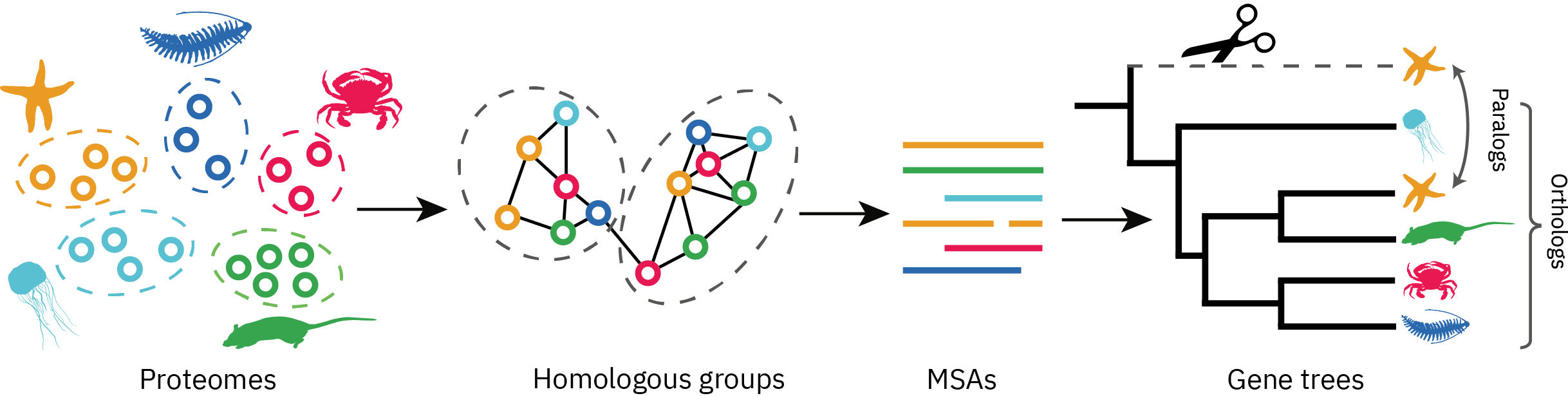
Figure 1. A rough overview of a tree-based orthology inference approach.
Quick installation
The easiest way to install PhyloPyPruner is by using the package manager for Python, pip:
pip install phylopypruner # install for all users
pip install --user phylopypruner # install for the current user only
Once installed, the program is located within $HOME/.local/bin. Depending on
your OS, you might have to add the directory to your $PATH to avoid typing
the entire path. Once in your path, you run the program like this:
phylopypruner
Documentation
Cite
Our manuscript is still in preparation, it will be posted here once a preprint of the article is available.
© Kocot Lab 2019
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file phylopypruner-1.3.2.tar.gz.
File metadata
- Download URL: phylopypruner-1.3.2.tar.gz
- Upload date:
- Size: 74.5 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.13.2
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
5d6ed1e6578aafa6ada6b15e24a3b0df3a4278189350696d8be030dfb54d317c
|
|
| MD5 |
7c000e07a293db2c602a89bf9814a358
|
|
| BLAKE2b-256 |
1690578616e19b61e6af2e117cf193a0d02f62685b1fd4933f2906b300b57d39
|
File details
Details for the file phylopypruner-1.3.2-py3-none-any.whl.
File metadata
- Download URL: phylopypruner-1.3.2-py3-none-any.whl
- Upload date:
- Size: 85.3 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.13.2
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
704206319a00b46d351075435d581d8635670cdf772acdc1aa38c9d270c5de6c
|
|
| MD5 |
d231dd55949d4b14bb63f14199b5cba4
|
|
| BLAKE2b-256 |
b59880b959ae65495d07f761110db2d97cd4ca9c9be3c83d4f92530af46aa92e
|







