A new profiling approach for DNA sequences based on the nucleotides' physicochemical features

Project description
A new profiling approach for DNA sequences based on the nucleotides' physicochemical features for accurate analysis of SARS-COV-2 genomes

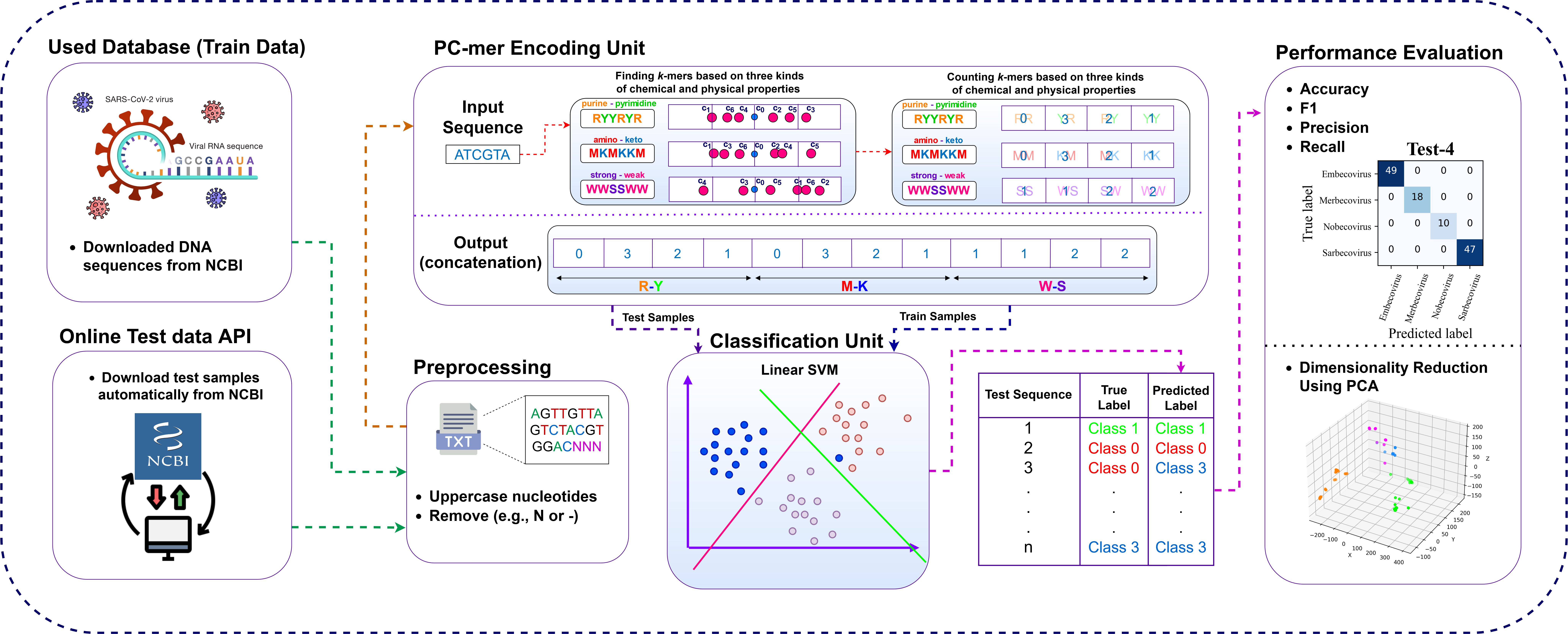
PC-mer Workflow
INTRODUCTION
The classification of different organisms into subtypes is one of the most important tools of organism studies, and among them, the classification of viruses itself has been the focus of many studies due to their use in virology and epidemiology. Many methods have been proposed to classify viruses, some of which are designed for a specific family of organisms and some of which are more general. But still, especially for certain categories such as Influenza and HIV, classification is facing performance challenges as well as processing and memory bottlenecks. In this way, we designed an automated classifier, called PC-mer, that is based on k-mer and physicochemical characteristics of nucleotides, which reduces the number of features about 2 k times compared to the alternative methods based on k-mer, and compared to integer and one-hot encoding methods, it is possible to keep the number of features constant despite the growth of the sequence length. In this way, it also increases the training speed by an average of 88%. This improvement in processing complexity is provided while PC-mer can also improve the classifying performance for a variety of virus families.
PC-mer's Performance
Classification Accuracy:
| Datasets | Accuracy (%) |
F1 (%) |
Precision (%) |
Recall (%) |
|---|---|---|---|---|
| Test-1 | 97.25 | 97.23 | 97.38 | 97.25 |
| Test-2 | 95.93 | 95.9 | 96.16 | 95.93 |
| Test-3a | 98.52 | 98.49 | 98.85 | 98.52 |
| Test-3b | 100 | 100 | 100 | 100 |
| Test-4 | 100 | 100 | 100 | 100 |
| Test-5 | 99.33 | 99.36 | 99.55 | 99.33 |
| Test-6 | 100 | 100 | 100 | 100 |
| Human Coronavirus | 100 | 100 | 100 | 100 |
Test Accuracy (Covid-19 Dataset)
| Datasets | Testing datasets | Prediction Accuracy (%) | Predicted label |
|---|---|---|---|
| Test-1 | 29 Covid-19 Virus sequences | 100 | Riboviria |
| Test-2 | 29 Covid-19 Virus sequences | 100 | Coronaviridae |
| Test-3(a/b) | 29 Covid-19 Virus sequences | 100 | Betacoronavirus |
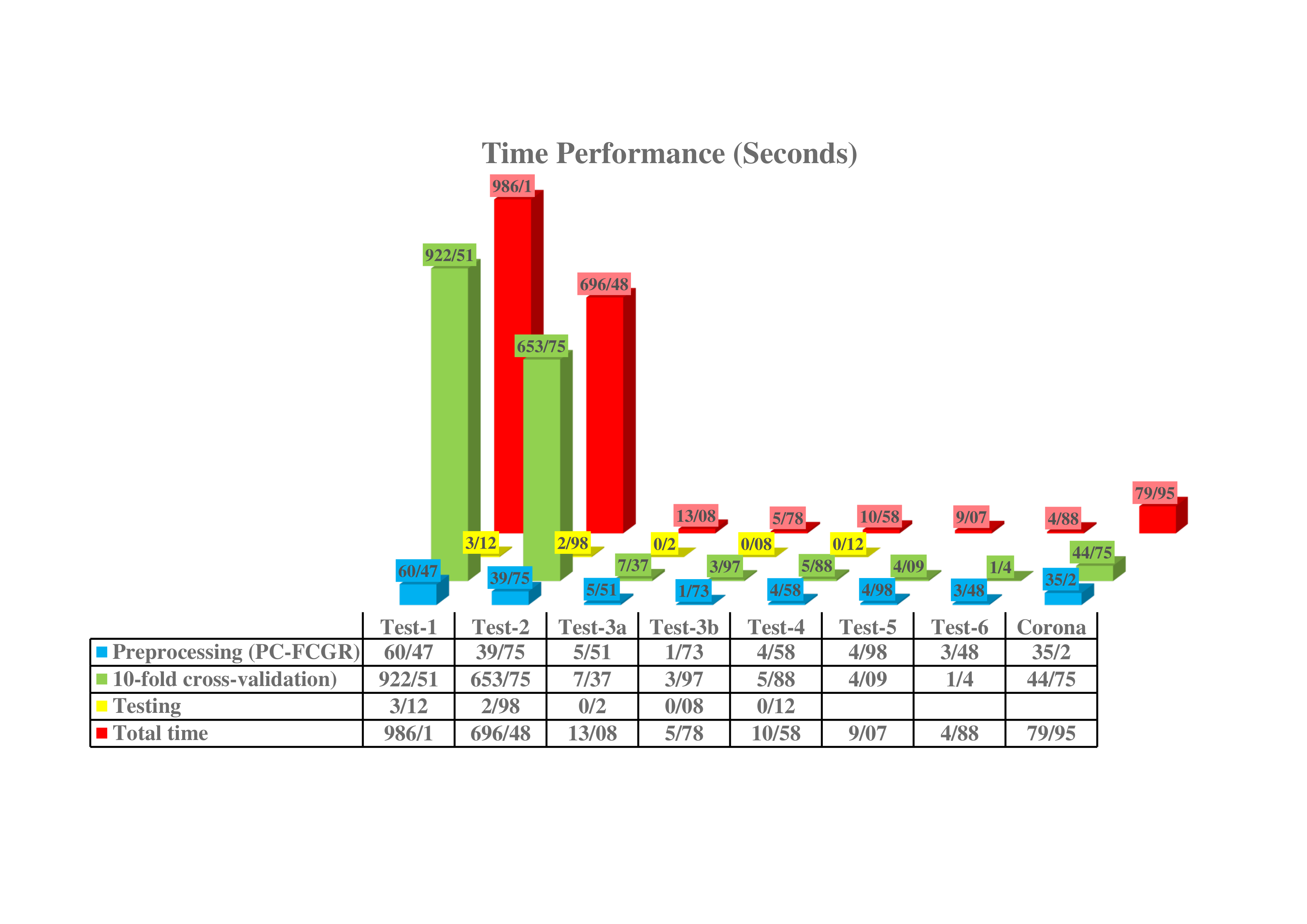
Time Performace:
As another advantage of our proposed encoding method, PC-mer can significantly improve the total processing time, which includes the runtimes of preprocessing, training, and testing procedures. Thanks to its powerful encoding algorithm and thus, facilitating usage of simple machine learning-based models to classify Coronaviridae family, all classification experiments, including preprocessing, training, and testing steps, have been performed on a desktop computer and a CPU processor.
Memory Consumption:
It is worth mentioning that PC-mer encoding allows usage of larger k-mers by reducing the size of encoded data. Specifically, PC-mer encoding is designed to reduce the computational overhead of k-mers, as well as the volume of the generated data from O(4^k) to O(3×2^k). For example, assuming k=7 in the FCGR method, a vector of size 16,384 is generated for each genome sequence, while PC-mer encoding generates a vector of size 12,288 for each genomic sequence, assuming k=12, which is much smaller than that of the FCGR’s generated data for k=7. It should be mentioned that assuming k-mers of size 12 in the FCGR method leads to a vector of size 16,777,216 per genome sequence. As a key superiority, PC-mer encoding with k = 7 (that produces vectors of size 384 for each genome sequence) achieves equal or higher accuracy, compared to the MLDSP tool with k-mers of size 12. We can conclude that the data compression achieved by the PC-mer encoding not only increases the classification accuracy and the feasibility of using larger k-mers, but also it leads to significant reduction in preprocessing, training, and testing times.
| Memory Consumption (PC-mer VS. FCGR) | Classification Accuracy for the variation of k-mer size in the range of [1,12] |
|---|---|
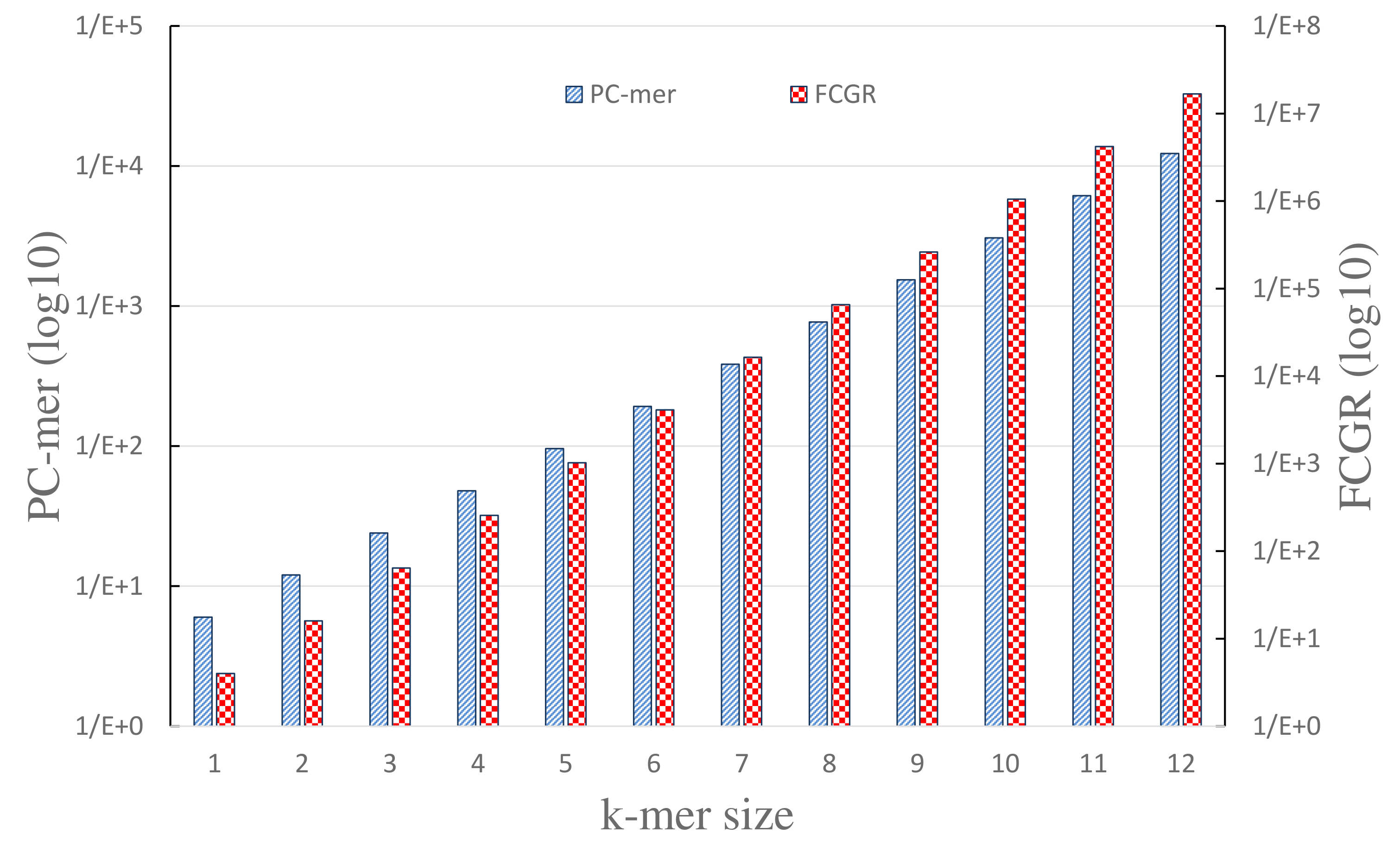 |
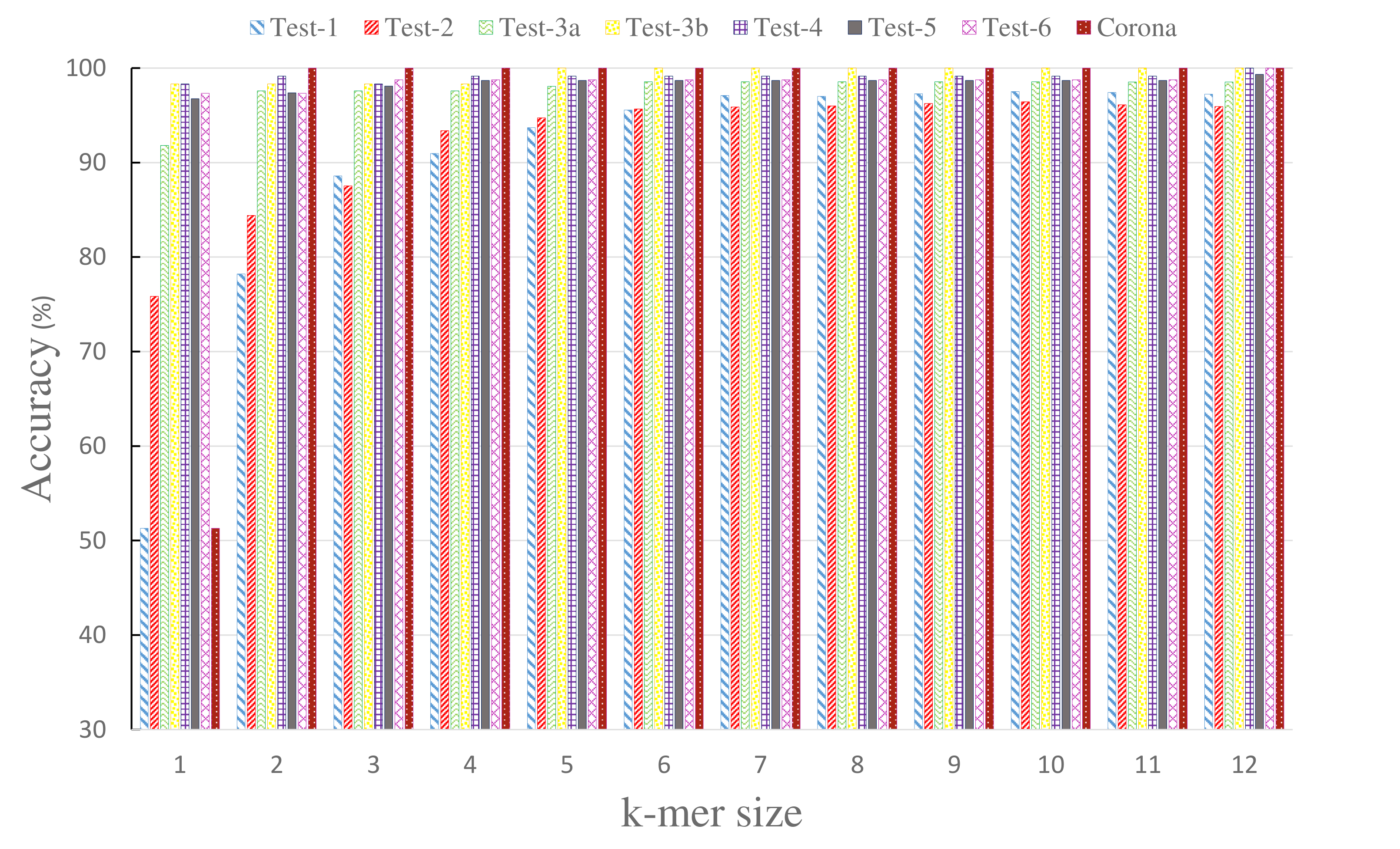 |
PREREQUISITES
The method was implemented in Python 3.8 with the use of scikit-learn library running on a normal desktop computer (CPU: i7-6500 2.5 GHz, RAM: 8 GB RAM, HD: 256GB Lexar, GPU: GeForce GTX 920M.
PC-mer Package
Main Features
Let's Take a Rapid Tour of PC-mer Functions:
Change_DNA(dna): This function takes the contents of a fasta file and extracts the nucleotides. Also, all nucleotides are replaced by capital letters.
#Example
>>> dna=">MN908947.3 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome\nATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGTAGATCTGTTCTCTAAA"
>>> Change_DNA(dna)
#Output:
'ATTAAAGGTTTATACCTTCCCAGGTAACAAACCAACCAACTTTCGATCTCTTGTAGATCTGTTCTCTAAA'
PC_mer(dna, k): k-mers generation function based on physicochemical properties. This function takes a sequence and sizekas input and its output is the desired feature vector.
#Example
>>> PC_mer(Seq,2)
#Output:
array([16, 14, 13, 27, 16, 15, 15, 24, 27, 18, 17, 8])
GFL(path,k): Takes apathand automatically reads all the fasta files in the desired path and returns the generatedfeature vectorsand theirlabels.
#Example
>>> GFL('data',2)
#Output:
([array([ 7327, 7490, 7489, 7597, 8887, 6570, 6570, 7876, 10780,
7768, 7767, 3588]), array([ 7315, 7490, 7489, 7597, 8887, 6570, 6570, 7864, 10768,
7768, 7767, 3588]), array([ 7291, 7469, 7468, 7574, 8861, 6627, 6627, 7687, 10391,
7817, 7816, 3778]), array([ 7257, 7453, 7452, 7570, 8793, 6638, 6638, 7663, 10390,
7799, 7798, 3745])], [0, 0, 1, 1])
train(PCmer_features, label, folds, clf): To evaluate the impact of encoding unit, we use six popular and practical supervised-learning based classification models (i.e. Logistic Regression (LR), Gaussian naïve Bayes (GNB), linear discriminant analysis (LDA), multi-layer perceptron (MLP), decision tree (DT), Linear Support Vector Classification (SVC)). So,Trainfunction is designed to classify encoded genomic sequences (PCmer_features) automatically. This function takesPCmer_features,Ground truth labels, the number of cross-validation operations (folds) and classification algorithm (clf). Classificationaccuracy metricsandrun timeare the outputs of train function.
#Example
>>> train(PCmer_features, label, 10,clf='LR')
#Output:
time = 0.09
accuracy = 1.000
precision = 1.000
recall = 1.000
f1 = 1.000
get_metrics(Groundtruth, Predicted): Calculation of accuracy metrics (F1,accuracy,recall,precision) based onGround truthandpredicted labels.
#Example
>>> actual = [1, 3, 3, 2, 5, 5, 3, 2, 1, 4, 3, 2, 1, 1, 2]
>>> predicted = [1, 2, 3, 4, 2, 3, 3, 2, 1, 2, 3, 1, 5, 1, 1]
>>> get_metrics(actual,predicted)
#Output:
(0.4266666666666667, 0.4666666666666667, 0.4444444444444444, 0.4666666666666667)
comp_confmat(Groundtruth, Predicted): Create a confusion matrix based onGround truthandpredicted labels.
#Example
>>> actual = [1, 3, 3, 2, 5, 5, 3, 2, 1, 4, 3, 2, 1, 1, 2]
>>> predicted = [1, 2, 3, 4, 2, 3, 3, 2, 1, 2, 3, 1, 5, 1, 1]
>>> comp_confmat(actual,predicted)
#Output:
array([[3., 0., 0., 0., 1.],
[2., 1., 0., 1., 0.],
[0., 1., 3., 0., 0.],
[0., 1., 0., 0., 0.],
[0., 1., 1., 0., 0.]])
plot_confusion_matrix(cm, target_names, title='Confusion matrix', cmap=None, normalize=True): This function displays theConfusion Matrixbased on comp_confmat function. The inputs of this function are cm (output of comp_confmat function) andtarget_names.
#Example
>>> plot_confusion_matrix(cm=CM,
normalize = False,
target_names = ['2019-nCoV','Sarbecovirus'],
title = "Test-6")
- pcmer_api(fname,seqid):
PCmer_apifunction downloads sequences from NCBI for training and testing PC-mer pipeline automatically. The inputs of this function are fname (your desired name for downloaded sample) andseqid.
#Example
>>> pcmer_api('sample-1','MG772933.1')
#Outputs:
Saved
Parsing...
ID: MG772933.1
Name: MG772933.1
Description: MG772933.1 Bat SARS-like coronavirus isolate bat-SL-CoVZC45, complete genome
Number of features: 0
Seq('ATATTAGGTTTTTACCTTCCCAGGTAACAAACCAACTAACTCTCGATCTCTTGT...AAA')
Now, generate your own pc-mer😉.
Quick Access Instructions
- Install:
!pip install physicochemical-mers==1.0.0
- Generate pcmer vectors:
from pcmer import features
#sample code
Seq = features.Change_DNA('id\nAGGAAAAGCCAACCAACCTCGATCTCTTGTAcct')
features = features.PC_mer(Seq, 2)
* A simple implementation *
Method's Limitations
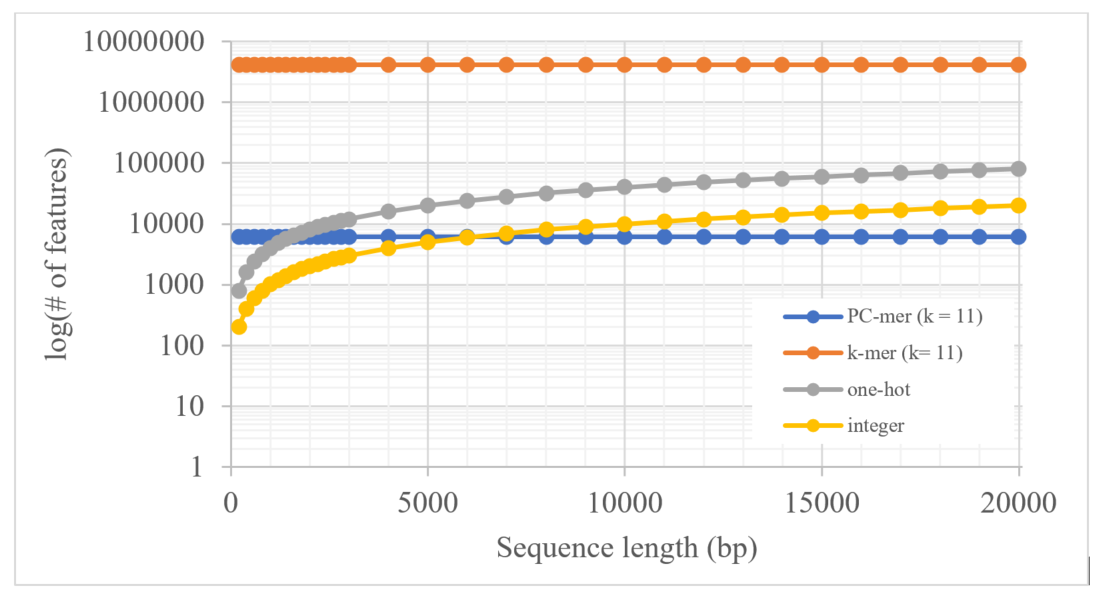
Length of Sequences: As mentioned, PC-mer is based on k-mer and physicochemical characteristics of nucleotides, which reduces the number of features about $2^k$ times compared to the alternative methods based on k-mer, and compared to integer and one-hot encoding methods, Also, PC-mer, similar to other k-mer-based methods, has the ability to convert sequences of any length into a fixed-length matrix. In this way, compared to methods such as integer and one-hot encoding, the number of extracted features remains constant. For more detailed comparison, the numbers of extracted features by the four methods (i.e. PC-mer (k = 11), k-mer (k = 11),Integer, andone-hot) are shown in below Figure for various sequence lengths. Although PC-mer can convert sequences of any length into a fixed-length matrix, its generalizability to achieve the acceptable accuracy for long sequences like humans is still a question. We achieved remarkable accuracy for the Coronavirus family with a maximum sequence length of50000, but it is an open problem for datasets with different abundance, number of categories, sequence length and taxonomic levels. Therefore, we would examine and deeply investigate thePC-merfor other datasets, as a stand-alone study, as our future work.see here for other viral genome classification based on PC-mer.
Acceptable Sequences: Most of the studies classifying whole-genome sequences either focus on noise-free data, or assume noise elimination as the preprocessing step. As a key capability of PC-mer, we also admitwhole genome sequencescontaining probable noises (nucleotides that cannot be determined definitively during sequencing), and remove these noises. Of course, there are other mechanisms, such as aligning the input sequences with their genome reference and making decisions about these letters according to the reference genome, which, of course, may not be definitive and depend on the amount of noises. In other words, these methods assume known input sequences, which is not necessarily the case in real problems. In other words, the input sequence may not be known, and so, consumers try to identify it with clustering and classification tools, so considering genome reference for aligning with it might be impossible. On the other hand, in the problems dealing with the whole-genome sequence, the ratio of noise to the length of the sequence is not so high to necessitate input alignment, while for those problems dealing with reads, such as metagenomics problems, the alignment solution is adopted. Of course, in the continuation of our studies on the PC-mer method and other data, we would also usereads, such asmetagenomicsproblems, and so, in future works,
CONTACT INFO
Somayyeh Koohi
Department of Computer Engineering
Sharif University of Technology
E-mail: koohi@sharfi.edu
Home page: http://sharif.ir/~koohi/
Release history Release notifications | RSS feed

Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file physicochemical-mers-1.0.0.tar.gz.
File metadata
- Download URL: physicochemical-mers-1.0.0.tar.gz
- Upload date:
- Size: 15.0 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.2 CPython/3.9.1
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d5d7609258191a8319d2d5ac093dabac50d20986ae51426cb45e9790dce347f1
|
|
| MD5 |
0c16cd5243651fc19662cf1bf8861a8b
|
|
| BLAKE2b-256 |
ea8465d2acba680ec141a6da82f3b8536194ed2149e01f685a52216929d55643
|
File details
Details for the file physicochemical_mers-1.0.0-py3-none-any.whl.
File metadata
- Download URL: physicochemical_mers-1.0.0-py3-none-any.whl
- Upload date:
- Size: 10.5 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.2 CPython/3.9.1
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
a8714a873e9936cc025a46ea295c287208c92969a2974cdec602492519ffe317
|
|
| MD5 |
692579803f8aa2029d06535213140050
|
|
| BLAKE2b-256 |
22c4f0427b0638eeba4d211c2b02f9e5acde0737bceb0f010843bc81c285fd16
|







