unsupervised classification of polyploid subgenomes

Project description
Quick summary
polyCRACKER is an unsupervised machine learning approach to the classification and extraction of sub-genomes from a set of genomic sequences provided in FASTA format. It currently tailored to the analysis of moderate to recently derived allopolyploid species. It does not require training data or even the number of subgenomes to be known (although this helps). It does require some empirical testing, however, in order to determine the most likely number of subgenomes.
polyCRACKER can be used to:
-
Identify subgenomes
-
Extract subgenomes
-
Validate subgenomes
-
Explorative analysis of subgenomes relative to genomic features
polyCRACKER works by using repeat kmers (corresponding to viruses, transposons, and other selfish repetitive elements) as molecular barcodes for identifying species of origin. Since such repetitive sequences evolve quickly and copy themselves throughout a genome of a species, but not other closely related species), they can be used to group subsequences based on species of origin.
Given a pool of DNA sequences derived from multiple species, polyCRACKER can be used to identify and separate sequences belonging to one species versus another. In some cases, polyCRACKER performs as well at separating subgenomes of an allopolyploid as the manual extraction of subgenomes by sequence alignment, when the progenitor genome sequences are known and available.
For more information, see the polyCRACKER manuscript preprint. Please cite the following article if you use polyCRACKER in your work.
(First and second authors are co-first authors.)
Getting Started With polyCRACKER
(requires mac or linux OS) (Docker works with Windows)
###Install of polyCRACKER dependencies
- Docker (recommended)
Install of dependencies can be skipped entirely by using the provided docker image available on github.
Miniconda-based image
The core functionality of polyCRACKER can be accessed by using a miniconda-based image:
docker pull sgordon/polycracker-miniconda:1.0.2
You may need to increase settings in docker to allow additional memory and CPU usage from within docker. Please see this thread: How to assign more memory to docker
We recommend allowing at least 5 Gb of RAM and at least 4 CPU.
Running polyCRACKER on test data
docker run -it sgordon/polycracker-miniconda:1.0.2
source activate pCRACKER_p27
tar -xzvf ./test_data/test_fasta_files/algae.fa.tar.gz && mv algae.fa ./test_data/test_fasta_files/
polycracker.py test_pipeline -env pCRACKER_p27
Results stored in test_results directory.
To exit the container:
exit
Note that if you want to inspect the results outside of the docker container, you may need to mount a volume.
The details on mounting a volume in the context of docker is outside the
scope of this tutorial. Nonetheless, if you have a analysis_results directory on your machine
and wish to copy the results from polyCRACKER to that directory, then you may modify the above commands to:
docker run -v "$(pwd)"/analysis_results:/analysis_results -i -t sgordon/polycracker-miniconda:1.0.2
source activate pCRACKER_p27
tar -xzvf ./test_data/test_fasta_files/algae.fa.tar.gz && mv algae.fa ./test_data/test_fasta_files/
polycracker.py test_pipeline -env pCRACKER_p27
cp -R test_results /analysis_results/
Then exit the container as above. The results should be persisted within the analysis_results/test_results subdirectory. You may also want to perform this mounting when running on your own data.
You may also build your own docker image by using the Dockerfile at the root of this repository. Details on this are described at the bottom of the page.
- Manual conda install of the required dependencies and run within a conda environment. See below for more details on manual conda installation of dependencies.
For more testing data:
- Tobacco (pseudomolecule-anchored and unanchored)
- 2017 Wheat genome
- Creinhardtii
- Csubellipsoidea
- Ustilago Ustma
- Ustilago Usthor
- Aspergillus species
Running on your own data using docker
- Edit config_polyCRACKER.txt (See below)
The flow will be similar to test data, but notably you will minimally need to: 2. Move fasta file in question to ./fasta_files This can be performed by mounting a volume to the docker container as described above, provided that the input FASTA file of interest is in the directory being mounted (for example, "analysis_results"), then copying the FASTA file from the mounted directory into the ./fasta_files directory that already exists within the container.
docker pull sgordon/polycracker-miniconda:1.0.2
# assumes we have copied user input FASTA file into analysis_results directory that we will mount
docker run -v "$(pwd)"/analysis_results:/analysis_results -i -t sgordon/polycracker-miniconda:1.0.2
source activate pCRACKER_p27
# copying user input FASTA file into fasta_files directory
cp /analysis_results/[user FASTA file] ./fasta_files
polycracker.py run_pipeline -env pCRACKER_p27
cp -R analysisOutputs /analysis_results/
Results should be in ./analysisOutputs/*/* sub-directories.
- There's a cluster results directory containing initial clusters of subsequences, and final results directory containing final clusters after signal amplification.
Sometimes signal amplification may fail due to the over agressive iterative recruitment of kmers that are either not subgenome-specific or they are specific to the opposite subgenome and incorrectly recruited. In this case, one can attain intermediate results by going into ./analysisOutputs/*/*/bootstrap_* directories and looking for extractedSubgenomes subdirectory containing fastas.
Note that extracted subgenome fasta files are still "chunked" (split according to the specified subsequence length during normalization), but contain positional information with respect to scaffold of origin.
-
Clustering plots found at in *html files in project directory.
-
Additional plots can be made using polycracker.py plotPositions -h, and there are a few other plotting utilities.
Pro tip: Can rerun/resume pipeline at various parts by setting parts of the config already run to 0 instead of 1.
Pro tip: Use command polycracker.py number_repeatmers_per_subsequence to find a histogram of the number of repeat-mers present in each chunked genome fragment. File saved as kmers_per_subsequence.png
If this histogram is too skewed to low kmer counts in each subsequence, then either:
- Reduce kmer size
- Increase chunk size splitFastaLineLength
- Reduce the low_count threshold
- Set perfectmode to 1
- Consider adding the NonChunk = 1 to config
- And/Or Enforce a higher MinChunkSize.
VERY IMPORTANT!
If there are is not enough repeat content included in the subsequences, they will be hard to bin. When running the pipeline, "kmers_per_subsequence.png" may be run in order to identify the frequency of kmers across subsequences and then tune relevant parameters.
Configuration for Running polyCRACKER Pipeline using Nextflow
polyCRACKER itself is a python module at the root of the repository and contains command line functions that can be individually accessed as noted above.
Because polyCRACKER consists of many individual commandline functions, we provide a pipeline written in nextflow workflow language for the convenience of users. The nextflow implementation allows a single command to then execute all the required steps in serial. This workflow is accessed for test data through:
polycracker.py test_pipeline
or as shown below for use on your own data:
polycracker.py run_pipeline.
The workflow itself is polyCRACKER_pipeline.nf, which is now within the polycracker subdirectory. Currently several resource parameters may need to be edited within the nextflow script itself, namely parameters on the number of CPU to use and memory resoures. These parameters are currently set to conservative values so that it may be run on test data on a modern laptop with 6 cores and at least 5 Gb of memory. When executing on larger datasets you will need to increase these resource settings. In particular, required memory resources scales with the size of the input FASTA sequences being analyzed. The parmeters can be change on these lines:
blastMemStr = "export _JAVA_OPTIONS='-Xms5G -Xmx" + blastMemory + "G'"
CPU requirements are specified in lines prefixed by "cpus" like this:
cpus = { writeKmer == 1 ? 6 : 1 }
polyCRACKER Configuration file setup
The provided config file within the root of the repository is 'config_polyCRACKER.txt'.
Parameters for controlling the amount of resources for individual functions and third party programs are set within the config file. Please modify as described below to suit your FASTA input as described below.
- File paths:
Copy your input FASTA file ( a single FASTA file with all your sequences ) into the
fasta_filesdirectory. You may alternatively modifyfastaPathto the path for your respective FASTA input file. You may leave the other paths as provided in the example config. FASTA files must end with.faor.fastafile extensions or they will not be recognized.
blastPath = ./blast_files/
kmercountPath = ./kmercount_files/
fastaPath = ./test_data/test_fasta_files/
bedPath = ./bed_files/
-
genome: The full filename (not the full path) of your input fasta file.
-
sge interpreter: Unless using an sge or slurm cluster, set local to 1. We do not currently document how to use sge or slurm muti-node clusters, but experienced users may try it on their own.
-
use bbtools: Please leave this set to 1.
-
Settings surrounding the number of anticipated subgenomes: Recommended practice, number of dimensions > number of subgenomes. Modify accordingly. For example, if the number of anticipated subgenomes is 2, then set n_dimensions to 3.
-
FASTA normalization Split FASTA into chunks. This determines the length of subsequences into which the input FASTA is divided into. This is necessary for normalizing subsequences for analysis. This is typically a value between 30000 and 1000000, but depends on the lengths of sequences within the input FASTA file. We recommend these as starting values:
splitFasta = 1
preFilter = 0
splitFastaLineLength = 50000
- Kmer counting settings 'kmerLength' is an important parameter that may need to be adjusted depending on the analysis. 'kmer_low_count', 'use_high_count', 'kmer_high_count' that control which kmers are used in the anaylsis. 'kmer_low_count' determines what kmers are considered 'repetitive'. 'use_high_count', 'kmer_high_count' limit the use of kmers found in the FASTA at high frequencies. We recommend these initial settings:
writeKmer = 1
kmerLength = 26
kmer2Fasta = 1
kmer_low_count = 30
use_high_count = 0
kmer_high_count = 2000000
sampling_sensitivity = 1
use original genome for final analysis output Typically this will be set to zero.
- **re-mapping kmers to the genome and transform of results into clustering matrix. specified memory usage options. 'blastMemory' is an important resource setting. Set this to the amount of RAM that you would like to use. On a laptop we recommend these settings:
writeBlast = 1
k_search_length = 13
runBlastParallel = 0
blastMemory = 5
blast2bed = 1
generateClusteringMatrix = 1
lowMemory = 0
minChunkSize = 50000
removeNonChunk = 1
minChunkThreshold = 0
tfidf = 1
perfect_mode = 0
On a larger single node cluster, you will want to increase the memory setting. 'removeNonChunk' excludes sequences shorter than the specified 'minChunkSize'.
transform and cluster the data: Two critical choices are what dimensionality reduction method to use and which cluster method to employ. 'reduction_techniques' indicates which method to use when performing dimensionality reduction on the sparse repeat-kmer by subsequence matrix. Available dimensionality reducers include:
- 'kpca': KernelPCA,
- 'factor': FactorAnalysis,
- 'feature': FeatureAgglomeration,
- 'lda': LatentDirichletAllocation, AND 'nmf': NMF.
Description of these methods is beyond the scope of this work.
'clusterMethods' specifies the cluster method that is used.
Supported methods are:
- 'SpectralClustering': SpectralClustering,
- 'hdbscan_genetic':GeneticHdbscan,
- 'KMeans': MiniBatchKMeans,
- 'GMM':GaussianMixture,
- 'BGMM':BayesianGaussianMixture.
Example parameters are:
transformData = 1
reduction_techniques = tsne
transformMetric = linear
ClusterAll = 1
clusterMethods = SpectralClustering
grabAllClusters = 1
n_neighbors = 20
metric = cosine
weighted_nn = 0
mst = 0
extract the subgenomes: Heuristics on subgenome repeat-kmer counts in order to say whether a subsequence belongs to one or another subgenome. Example parameters:
extract = 0
diff_kmer_threshold = 20
default_kmercount_value = 3
diff_sample_rate = 1
unionbed_threshold = 10,2
bootstrap = 1
Using polyCRACKER command line functions outside the automated nextflow pipeline.
polyCRACKER is a python module with command line accessible functions. The nextflow run pipeline scripts allow users to avoid the need to run individual functions in serial for the common purpose of subgenome classification and extraction.
Nonetheless there are instances where execution of individual core and helper functions are useful.
To see the full list of command line available functions:
docker pull sgordon/polycracker-miniconda:1.0.2
docker run -it sgordon/polycracker-miniconda:1.0.2
source activate pCRACKER_p27
polycracker.py -h
The resulting list:
Usage: polycracker.py [OPTIONS] COMMAND [ARGS]...
Options:
--version Show the version and exit.
-h, --help Show this message and exit.
Commands:
TE_Cluster_Analysis Build clustering matrix (repeat counts vs...
align Align two fasta files.
anchor2bed Convert syntenic blocks of genes to bed...
avg-distance-between-diff-kmers
Report the average distance between...
bed2scaffolds-pickle Convert correspondence bed file,...
bio-hyp-class Generate count matrix of kmers versus...
blast2bed Converts the blast results from blast or...
blast_kmers Maps kmers fasta file to a reference...
build-pairwise-align-similarity-structure
Take consensus repeats, generate graph...
categorize-repeats Take outputs from denovo repeat...
cluster Perform clustering on the dimensionality...
cluster-exchange Prior to subgenome Extraction, can choose...
clusterGraph Plots nearest neighbors graph in html...
color-trees Color phylogenetic trees by progenitor of...
compare-scimm-metabat
compare-subclasses In development: Grab abundance of top...
compareSubgenomes_progenitors_v_extracted
Compares the results found from the...
convert-mat2r Convert any sparse matrix into a format...
convert_subgenome_output_to_pickle
Find cluster labels for all...
correct-kappa Find corrected cohen's kappa score.
count-repetitive Infer percent of repetitive sequence in...
dash-genetic-algorithm-hdbscan-test
Save labels of all hdbscan runs, generate...
dash-genome-quality-assessment Input pre chromosome level scaffolded...
diff-kmer-analysis Runs robust differential kmer analysis...
differential_TE_histogram Compare the ratio of hits of certain...
explore-kmers Perform dimensionality reduction on...
extract-scaffolds-fasta Extract scaffolds from fasta file using...
extract-sequences Extract sequences from fasta file and...
final_stats Analyzes the accuracy and agreement...
find-best-cluster-parameters In development: Experimenting with...
find-denovo-repeats Wrapper for repeat modeler.
find-rules In development: Elucidate underlying...
find-rules2 In development: Elucidate underlying...
generate-genome-signatures Wrapper for sourmash.
generate-karyotype Generate karyotype shinyCircos/omicCircos...
generate-out-bed Find cluster labels for all...
generate-unionbed Generate a unionbedgraph with intervals...
generate_Kmer_Matrix From blasted bed file, where kmers were...
genomic-label-propagation Extend polyCRACKER labels up and...
get-density Return gene or repeat density information...
hipmer-output-to-kcount Converts hipmer kmer count output into a...
kcount-hist Outputs a histogram plot of a given kmer...
kcount-hist-old Outputs a histogram plot of a given kmer...
kmer2Fasta Converts kmer count file into a fasta...
kmerratio2scaffasta Bin genome regions into corresponding...
link2color Add color information to link file for...
maf2bed Convert maf file to bed and perform stats...
mash-test Sourmash integration in development.
merge-split-kmer-clusters In development: working on merging and...
multicol2multifiles Take matrix of total differential kmer...
number_repeatmers_per_subsequence
Find histogram depicting number of repeat...
out-bed-to-circos-csv Take progenitor mapped, species ground...
plot-distance-matrix Perform dimensionality reduction on...
plot-rules Plots normalized frequency distribution...
plot-rules-chromosomes Plot distribution of rules/conservation...
plot-unionbed Plot results of union bed file, the...
plotPositions Another plotting function without...
polyploid-diff-kmer-comparison Compare highly informative differential...
progenitorMapping Takes reference progenitor fasta files,...
repeat-subclass-analysis Input repeat_fasta and find phylogenies...
reset-cluster Delete cluster results, subgenome...
reset-transform Remove all html files from main work...
return-dash-data-structures Return dash data structures needed to run...
run-iqtree Perform multiple sequence alignment on...
run-metabat
run-tests Run basic polyCRACKER tests to see if...
run_pipeline Run polyCRACKER pipeline locally or on...
scaffolds2colors-specified Attach labels to each scaffold for use of...
send-repeats Use bbSketch to send fasta file...
shiny2omic Convert shinyCircos csv input files to...
species-comparison-scaffold2colors
Generate color pickle file for...
spectral-embed-plot Spectrally embed PCA data of any origin.
splitFasta Split fasta file into chunks of a...
subgenome-extraction-via-repeats
Extends results of TE_cluster_analysis by...
subgenomeExtraction Extract subgenomes from genome, either...
test_pipeline
transform_plot Perform dimensionality reduction on a...
txt2fasta Extract subgenome fastas from reference...
unionbed2matrix Convert unionbed file into a matrix of...
update_nextflow_config Updates nextflow configuration file for...
writeKmerCount Takes list of fasta files and runs...
To obtain information on a specific function, for example, plotPositions:
docker pull sgordon/polycracker-miniconda:1.0.2
docker run -it sgordon/polycracker-miniconda:1.0.2
source activate pCRACKER_p27
polycracker.py plotPositions -h
result for the above:
Usage: polycracker.py plotPositions [OPTIONS]
Another plotting function without emphasis on plotting the spectral graph. Emphasis
is on plotting positions and clusters.
Options:
-npy, --positions_npy PATH If standard layout, then use these data points to
begin simulation. [default:
graphInitialPositions.npy]
-p, --labels_pickle PATH Pickle file containing scaffolds. [default:
scaffolds.p]
-c, --colors_pickle PATH Pickle file containing the cluster/class each
label/scaffold belongs to. [default: colors_pickle.p]
-o, --output_fname PATH Desired output plot name in html. [default:
output.html]
-npz, --graph_file PATH Sparse nearest neighbors graph npz file. If desired,
try spectralGraph.npz.
-l, --layout [standard|spectral|random]
Layout from which to plot graph. [default: standard]
-i, --iterations INTEGER Number of iterations you would like to simulate to. No
comma delimit, will only output a single iteration.
[default: 0]
-s, --graph_sampling_speed INTEGER
When exporting the graph edges to CSV, can choose to
decrease the number of edges for pdf report
generation. [default: 1]
-ax, --axes_off When enabled, exports graph without the axes.
-cmap, --new_colors TEXT Comma delimited list of colors if you want control
over coloring scheme. [default: ]
-h, --help Show this message and exit.
Re-running subgenome classification and extraction during manual optimization
Two functions of immediate interest in the context of manual optimization and trouble shooting are:
polycracker.py reset-cluster -h
result:
Usage: polycracker.py reset-cluster [OPTIONS]
Delete cluster results, subgenome extraction results and corresponding html files.
and
polycracker.py reset-transform -h
result:
Usage: polycracker.py reset-transform [OPTIONS]
Remove all html files from main work directory. Must do this if hoping to
retransform sparse data.
The above functions remove some intermediate files as required to be able to successfully re-run the pipeline.
Additional Documentation
Other tips on setting up the config file and running the pipeline are found by running the jupyter notebook ./tutorials/RunningPipeline.ipynb
* Information on what each config parameter means is in this notebook. Highly recommend that you check this out.
* Other examples of old configuration files in ./tutorials/old_configs
Other downstream analyses not included here, but check out the html file described below for more commands.
Accessing additional help docs:
* You can find them here after you download the repository: ./tutorials/help_docs/index.html
* This is an html file that specifies some of the polyCRACKER commands. Still being updated.
Genome Comparison Tool and K-Mer Conservation Rules
- A separate utility of polyCRACKER that is NOT demonstrated in the paper above is the ability to compare the distribution of k-mers between different genomes/assemblies, and create a plotly/dash app for visualization.
- To establish a matrix of k-mers versus genomes for downstream analysis, please use bio_hyp_class command (-h)
* Eg. nohup python polycracker.py bio_hyp_class -f ../../,_,n -dk 5 -w ../../results/ -m 150 -l 23 -min 2 -max 25 > ../../analysis.log & - There are then scripts that can be used for downstream analysis (clustering, etc. not detailed here). This aspect will be published in a separate manuscript, in preparation.
Detailed instructions for environment setup
Building your own docker image
(from the provided Dockerfile at the root of this repository.)
The Dockerfile tested should build and run successfully in its current state. To build the image:
docker build . -t polycracker
docker run -it polycracker
source activate pCRACKER_p27
The recipe for conda install of the polyCRACKER environment
(Note that the Docker method is preferred and much easier.)
conda create -y --name pCRACKER_p27 python=2.7
conda activate pCRACKER_p27
conda install -y -c bioconda nextflow scipy pybedtools pyfaidx pandas numpy bbmap
conda install -y -c anaconda networkx click biopython matplotlib scikit-learn seaborn pyamg
conda install -y -c plotly plotly
conda install -y -c conda-forge deap hdbscan multicore-tsne
Test your conda environment by running polyCRACKER to classify algae genomes
- Clone the repository to your project directory.
git clone git@bitbucket.org:berkeleylab/jgi-polycracker.git
- change cd [your root of the git project directory containing polycracker.py]
cd [your project directory containing polycracker.py]
- tar -xzvf ./test_data/test_fasta_files/algae.fa.tar.gz && mv algae.fa ./test_data/test_fasta_files/
- Activate your conda environment
source activate pCRACKER_p27
- polycracker.py test_pipeline -env [Your polyCRACKER conda environment]. For example:
polycracker.py test_pipeline -env pCRACKER_p27
- Results stored in test_results directory.
Gallery
Example Plots
Deconvolution of green alga genomes Coccomyxa subellipsoidea and Chlamydomonas reinhardtii
(Plots result of spectral embedding of dimensionality reduced repeat-kmer matrix, Genomes split into 50kb subsequences before classification.)
Assigning sequences in the large tetraploid tobacco genome into two progenitor subgenomes
Classification of sequences in the massive hexaploid bread wheat genome into three ancestral subgenomes
Schematics
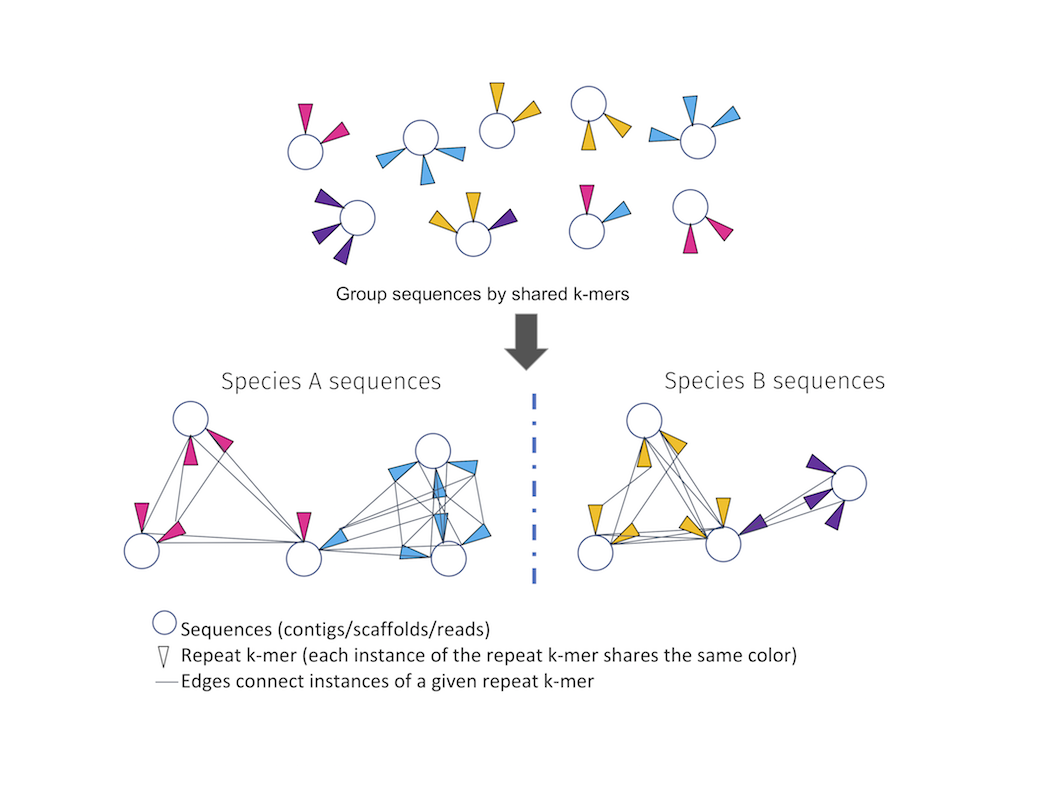
Illustrative schematic of polyCRACKER clustering of sequences linked by the repeat-kmers that they contain


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file polycracker-1.0.3.tar.gz.
File metadata
- Download URL: polycracker-1.0.3.tar.gz
- Upload date:
- Size: 117.8 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/1.11.0 pkginfo/1.5.0.1 requests/2.22.0 setuptools/41.0.1 requests-toolbelt/0.8.0 tqdm/4.23.3 CPython/2.7.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
b027824a661b17180271d70974a7a844a7abb36440176b51f90cf29591508d7f
|
|
| MD5 |
89beaa4df1d40981f0e1af8bdb09350f
|
|
| BLAKE2b-256 |
1cba8008da28a83dc55f85734c97a8b941fc21bd2a931ecf3dbdf23942d193f9
|
File details
Details for the file polycracker-1.0.3-py2-none-any.whl.
File metadata
- Download URL: polycracker-1.0.3-py2-none-any.whl
- Upload date:
- Size: 104.4 kB
- Tags: Python 2
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/1.11.0 pkginfo/1.5.0.1 requests/2.22.0 setuptools/41.0.1 requests-toolbelt/0.8.0 tqdm/4.23.3 CPython/2.7.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
80d47c9b5d8a49709ebacb4b95b6477035072676838c2e0bc5bdf777681a539d
|
|
| MD5 |
57499112e36c610da10bc6dc976f2b1a
|
|
| BLAKE2b-256 |
7ff2b4e0141ffd3a3fb6de16a1ddbfba117f589e62478567063002b352811457
|







