A versatile sequence read processor for nanopore direct RNA sequencing

Project description
Poreplex
Signal-level preprocessor for Oxford Nanopore direct RNA sequencing (DRS) data. Poreplex does many preprocessing steps required before the downstream analyses for RNA Biology and yields the processed data in the ready-to-use forms.




Features
- Demultiplexing barcoded direct RNA sequencing libraries
- Trimming 3′ adapter sequences
- Filtering pseudo-fusion reads
- Basecalling with ONT albacore (even faster than albacore itself)
- Live basecalling and processing during the sequencing
- Real-time read alignments with minimap2
- Full-screen dashboard view for real-time reports
- Various ready-to-use output formats
Installation
Poreplex requires Python 3.5+ and pip to install. This pip command installs poreplex with its essential dependencies. You may use the following command.
pip install poreplex
To install it together with all optional dependencies (except albacore), use this command:
pip install 'poreplex[full]'
Additional (Optional) Dependency
As its inputs, poreplex requires the FAST5 files that were basecalled using ONT albacore in advance. Alternatively, poreplex can also internally call albacore during the processing without a prior basecalling if the albacore package is available from the environment.
Quick Start
Produce FASTQ files without 3′ adapter sequences from a bunch of FAST5 files.
poreplex -i path/to/fast5 -o path/to/output --trim-adapter
Four direct RNA sequencing libraries can be barcoded, pooled and sequenced together. Porplex can demultiplex the librariess into separate FASTQ files.
poreplex -i path/to/fast5 -o path/to/output --trim-adapter --barcoding
In addition, poreplex can create directories containing hard-links to the original FAST5 files, organized separately by the barcodes.
poreplex -i path/to/fast5 -o path/to/output --trim-adapter --barcoding --fast5
In case the FAST5 files are not basecalled yet, just a switch lets poreplex call albacore internally. Multicore machines help.
poreplex -i path/to/fast5 -o path/to/output --trim-adapter --barcoding --fast5 --basecall --parallel 40
With the --live switch, All tasks can be processed as soon as reads
are produced from MinKNOW.
poreplex -i path/to/fast5 -o path/to/output --trim-adapter --barcoding --basecall --parallel 40 --live
One may want to output aligned reads directly to BAM files instead of FASTQ outputs. Poreplex streams the processed reads to minimap2 and update the BAM outputs real-time. A pre-built index (not a FASTA) generated using minimap2 must be provided for this.
poreplex -i path/to/fast5 -o path/to/output --trim-adapter --barcoding --basecall \
--parallel 40 --live --align GRCz11-transcriptome.mmidx
More vibrant feedback is provided if you turn on the dashboard switch.
poreplex -i path/to/fast5 -o path/to/output --trim-adapter --barcoding --basecall \
--parallel 40 --live --align GRCz11-transcriptome.mmidx --dashboard
Poreplex detects pseudo-fusion reads which may originate from
insufficiently segmented signals if --filter-chimera is given. This
improves the overall accuracy of demultiplexing.
poreplex -i path/to/fast5 -o path/to/output --filter-chimera
Barcoding direct RNA sequencing libraries
The official kits and protocols do not support barcoding in the direct RNA sequencing yet. Poreplex enables pooling multiple libraries into a single DRS run.
ONT direct RNA sequencing libraries are prepared by subsequently attaching two different 3' adapters, RTA and RMX, respectively. Both are double-stranded DNAs with Y-burged ends on the 3'-sides. Barcoded libraries for poreplex can be built with modified versions of RTA adapters. Unlike in the DNA sequencing libraries, poreplex demultiplexes in signal-level to ensure the highest accuracy. The poreplex package comes with pre-trained demultiplexer models for four different DNA barcodes. Order these sequences as many as you need in the experiment and replace the original RTA adapters.
BC1 Oligo A: 5'-/5Phos/CCTCCCCTAAAAACGAGCCGCATTTGCGTAGTAGGTTC-3'
BC1 Oligo B: 5'-GAGGCGAGCGGTCAATTTTCGCAAATGCGGCTCGTTTTTAGGGGAGGTTTTTTTTTT-3'
BC2 Oligo A: 5'-/5Phos/CCTCGTCGGTTCTAGGCATCGCGTATGCTAGTAGGTTC-3'
BC2 Oligo B: 5'-GAGGCGAGCGGTCAATTTTGCATACGCGATGCCTAGAACCGACGAGGTTTTTTTTTT-3'
BC3 Oligo A: 5'-/5Phos/CCTCCCACTTTCACACGCACTAACCAGGTAGTAGGTTC-3'
BC3 Oligo B: 5'-GAGGCGAGCGGTCAATTTTCCTGGTTAGTGCGTGTGAAAGTGGGAGGTTTTTTTTTT-3'
BC4 Oligo A: 5'-/5Phos/CCTCCTTCAGAAGAGGGTCGCTTCTACCTAGTAGGTTC-3'
BC4 Oligo B: 5'-GAGGCGAGCGGTCAATTTTGGTAGAAGCGACCCTCTTCTGAAGGAGGTTTTTTTTTT-3'
Basecalling with the ONT Albacore
Most studies requiring signal-level analysis need re-basecalling with the ONT albacore to get the event tables in the FAST5 files. Poreplex can internally call the basecaller core routines of albacore to yield the sequences and tables for the downstream analyses. In fact, running albacore via poreplex is remarkably faster than running albacore itself in a multi-core machine thanks to more efficient scheduling of the computational loads.
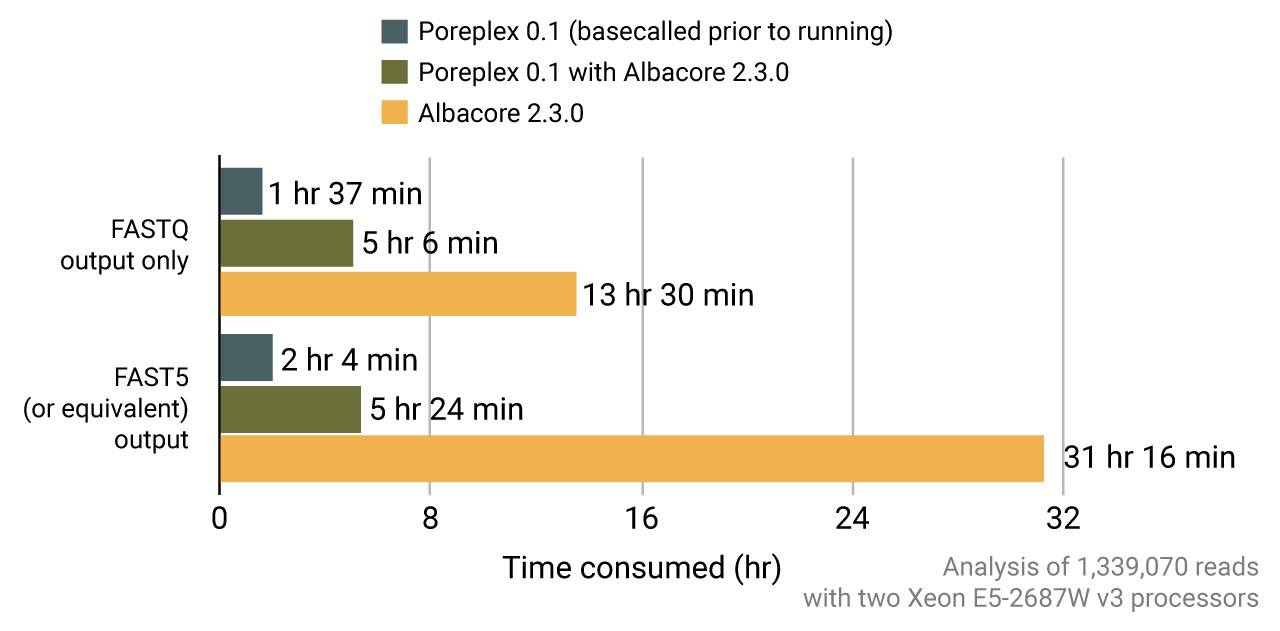
Live basecalling and processing
One can start the poreplex pipeline at any time even before the
sequencing begins. With the --live switch turned on, it monitors
every update in input directories and picks the newly created
files up for the whole process of the analysis. In the live mode,
the program keeps running unless a user presses Ctrl-C (in the
standard progress view) or Q (in the full-screen dashboard view). The
inotify module is required to allow
poreplex to run in the live mode.
In case the points of sequencing and analysis are different, a real-time directory synchronization software like DirSync Pro may help. Poreplex detects new files introduced by moving or closing a file after writing. Files that are made visible by creating hard or symbolic links or changing permissions may remain undetected.
Real-time sequence alignments
Poreplex aligns the reads to a reference transcriptome using minimap2 and writes the results to BAM files when an index file for the reference is provided. Some options that affect the performance of the alignments can be specified when generating the minimap2 index.
wget 'ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_28/gencode.v28.transcripts.fa.gz'
minimap2 -H -k 13 -w 10 -d gencode.v28.transcripts.mmidx gencode.v28.transcripts.fa.gz
poreplex -i /path/to/input -o /path/output --basecall --align gencode.v28.transcripts.mmidx
By default, switching on the alignments suppresses the FASTQ
outputs. Those can be recovered by adding --fastq to the command line.
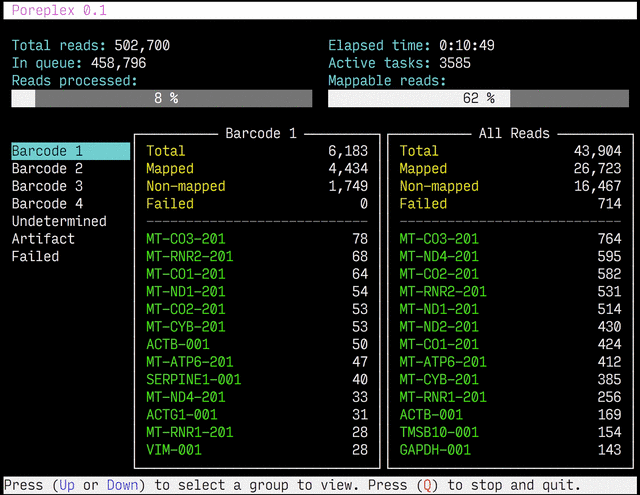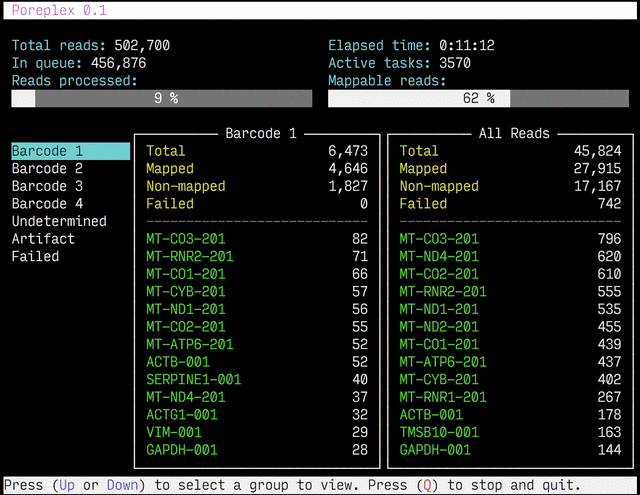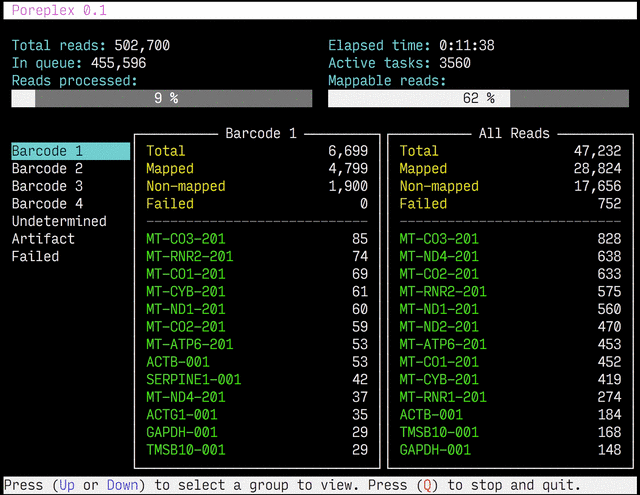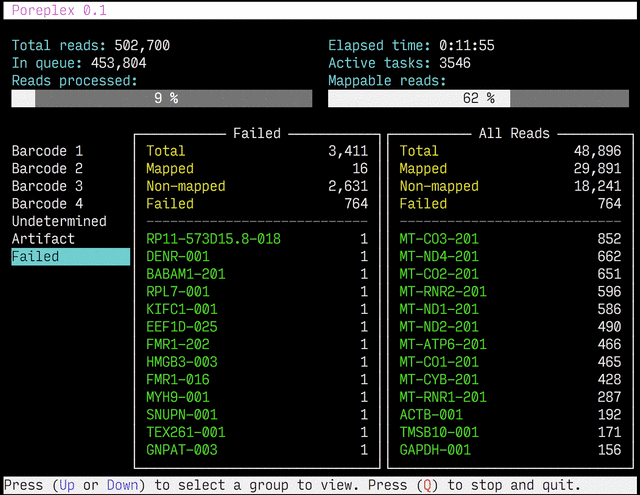
Real-time reports
The results from the real-time alignments with the overall progression of
the pipeline can be visualized as a full-screen dashboard view in a text
terminal. Poreplex shows the real-time report when the
command line includes the --dashboard switch along with --align for
the index of the reference transcriptome. The names of mapped sequences
are shown as the sequence name in the reference minimap2 index. To
see them as more familiar names, supply a file containing IDs and names
with the --contig-aliases switch. It must be a tab-separated
text file with two columns. The first should contain IDs (in the reference index)
and the second should contain names (to show in the screen). The read counts
window in the middle of the screen represents the summary of reads categorized by
error status or detected barcodes for multiplexed libraries. A user can
choose a group to show in the window with up and down arrow keys. To stop the
process and close the dashboard, press Q key at any time.
Pseudo-fusion filter
In the Oxford Nanopore strand sequencing, a read is a snippet from a very long contiguous signal from a channel. In most cases, there is a gap between two different molecules. The gap should be long enough for the MinKNOW to cut signals at the end of sequences. However, the gap between strands is sometimes not enough, so that a small fraction of reads carry two or more molecules. This phenomenon can be particularly problematic in the pooled libraries with barcodes and fusion gene studies. In a few runs in our testing, up to 1% of reads could be derived from insufficiently segmented signals. The following plot shows a signal sequence continued without any gap between the ends of two differently adapted RNAs which were prepared independently until the second ligation step, before which RNAs were pooled (RT before the ligation includes the heat- inactivation of enzymes).
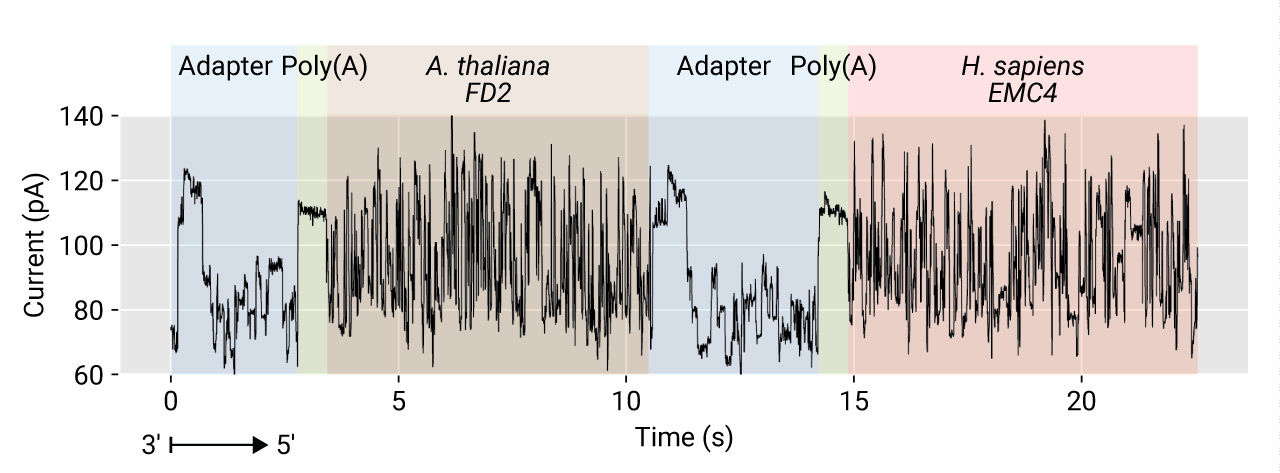
Poreplex detects potential artifacts by detecting multiple appearances of the
signature of the DNA adapter in a single read when the --filter-chimera switch
is turned on. Note that the default parameters for the filtering can be
too sensitive for some experiments.
Output formats
FASTQ
Sequences and quality scores are written to bgzip-compressed
FASTQ files in the fastq subdirectory. Each FASTQ file contains the
entire sequences of a group classified by processing status and
detected barcode.
| File name | Description |
|---|---|
fastq/pass.fastq.gz |
All sequences that were basecalled and passed the basic quality filters in poreplex. With --barcoding, the passed sequences with no identifiable barcode are written to this file. |
fastq/BC#.fastq.gz |
Sequences with identifiable barcode signals. |
fastq/fail.fastq.gz |
Too short sequences that could not be calibrated for the signal processing. |
fastq/artifact.fastq.gz |
Sequences that were classified as potential artifacts. |
FASTQ outputs are suppressed when BAM outputs are activated with
the --align option. Please add --fastq to restore the FASTQ outputs.
FAST5
To reduce the disk I/O, poreplex utilizes the links instead of copying
the original FAST5 to append basecalled results to the file. With the
--fast5 option, poreplex creates hard links of the original FAST5
files reorganized in subdirectories representing each processing status
or barcode. Symbolic links are created in case the hard links are not
possible or --symlink-fast5 is specified.
The basecalled events, which are stored in Analyses/Basecall_1D_00*
of the standard FAST5 files, are written to the events subdirectory
instead upon request by --dump-basecalled-events. The basecall
event tables for all reads are accessible through a single HDF5 file,
events/inventory.h5, by the read id. These tables include an
additional scaled_mean column, which contains mean current levels
scaled to match the ONT's reference
pore model.
BAM
The alignments to the reference transcriptome go into BAM files
inside the bam subdirectory. The reference sequences must be
indexed using minimap2 before providing it with the --align
option (see above). The BAM
files are not sorted and not filtered thoroughly. FASTQ or FASTA
sequence files can be generated from the BAM files without loss
using bedtools.
Please use these sequence alignments in the BAM files for quality
checks and sketchy analyses only.
Nanopolish database
Nanopolish provides very convenient
tools that help signal-level analyses. Poreplex provides a set of index
files that are required to run the nanopolish commands. Add --nanopolish
to a poreplex command line, then just skip nanopolish extract or
nanopolish index commands in its tutorial, and proceed directly to
the main steps.
Command line options
usage: poreplex -i DIR -o DIR [-c NAME] [--trim-adapter]
[--minimum-length LEN] [--filter-chimera] [--barcoding]
[--polya] [--basecall] [--align INDEXFILE] [--live]
[--live-delay SECONDS] [--fastq] [--fast5] [--symlink-fast5]
[--nanopolish] [--dump-adapter-signals]
[--dump-basecalled-events] [--dashboard]
[--contig-aliases FILE] [-q] [-y] [-p COUNT] [--tmpdir DIR]
[--batch-size SIZE] [--version] [-h]
| Short option | Long option | Description |
|---|---|---|
| Data Settings | ||
-i DIR |
--input DIR |
path to the directory with the input FAST5 files (required) |
-o DIR |
--output DIR |
output directory path (Required) |
-c NAME |
--config NAME |
path to signal processing configuration |
| Basic Processing Options | ||
--trim-adapter |
trim 3′ adapter sequences from FASTQ outputs | |
--filter-chimera |
remove unsplit reads fused of two or more RNAs in output | |
| Optional Analyses | ||
--barcoding |
sort barcoded reads into separate outputs | |
--polya |
output poly(A) tail length measurements | |
--basecall |
call the ONT albacore for basecalling on-the-fly | |
--align INDEXFILE |
align basecalled reads using minimap2 and create BAM files | |
| Live Mode | ||
--live |
monitor new files in the input directory | |
--live-delay SECONDS |
time to delay the start of analysis in live mode (default: 60) | |
| Output Options | ||
--fastq |
write to FASTQ files even when BAM files are produced | |
--fast5 |
link or copy FAST5 files to separate output directories | |
--symlink-fast5 |
create symbolic links to FAST5 files in output directories even when hard linking is possible | |
--nanopolish |
create a nanopolish readdb to enable access from nanopolish | |
--dump-adapter-signals |
dump adapter signal dumps for training | |
--dump-basecalled-events |
dump basecalled events to the output | |
| User Interface | ||
--dashboard |
show the full screen dashboard | |
--contig-aliases FILE |
path to a tab-separated text file for aliases to show as a contig names in the dashboard (see README) | |
-q |
--quiet |
suppress non-error messages |
-y |
--yes |
suppress all questions |
| Pipeline Options | ||
-p COUNT |
--parallel COUNT |
number of worker processes (default: 1) |
--tmpdir DIR |
temporary directory for intermediate data | |
--batch-size SIZE |
number of files in a single batch (default: 128) | |
--version |
show program's version number and exit | |
-h |
--help |
show this help message and exit |
Citing Poreplex
A pre-print is going to be uploaded soon.


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distributions
Built Distributions
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file poreplex-0.5-cp37-cp37m-manylinux1_x86_64.whl.
File metadata
- Download URL: poreplex-0.5-cp37-cp37m-manylinux1_x86_64.whl
- Upload date:
- Size: 942.8 kB
- Tags: CPython 3.7m
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/2.0.0 pkginfo/1.5.0.1 requests/2.21.0 setuptools/41.2.0 requests-toolbelt/0.9.1 tqdm/4.31.1 CPython/3.6.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
c4439351df5d261df8cd64b52266842ab68254af83d5ce712099a3203e1794db
|
|
| MD5 |
85cf1f0185dc38e2ed4a3b981bbe95ea
|
|
| BLAKE2b-256 |
dbb0986fd950bfce4a166f9aae554de7b77525bfcb0f5fcba8a3c4808f11185c
|
File details
Details for the file poreplex-0.5-cp36-cp36m-manylinux1_x86_64.whl.
File metadata
- Download URL: poreplex-0.5-cp36-cp36m-manylinux1_x86_64.whl
- Upload date:
- Size: 941.8 kB
- Tags: CPython 3.6m
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/2.0.0 pkginfo/1.5.0.1 requests/2.21.0 setuptools/41.2.0 requests-toolbelt/0.9.1 tqdm/4.31.1 CPython/3.6.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d362bda52bea7664176e51f214f5e5da299a367575051339ac3bf6b095dc9fe4
|
|
| MD5 |
a08b996dc47021286ed98482d9d91242
|
|
| BLAKE2b-256 |
9a954186abdb42830b0907afeed9c95d54f2a8708e6485edcaaadbbcc5bc51bd
|
File details
Details for the file poreplex-0.5-cp35-cp35m-manylinux1_x86_64.whl.
File metadata
- Download URL: poreplex-0.5-cp35-cp35m-manylinux1_x86_64.whl
- Upload date:
- Size: 941.7 kB
- Tags: CPython 3.5m
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/2.0.0 pkginfo/1.5.0.1 requests/2.21.0 setuptools/41.2.0 requests-toolbelt/0.9.1 tqdm/4.31.1 CPython/3.6.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
de86590bebc549000cbcec8346bfb625d58d545f609692716b254311c3630112
|
|
| MD5 |
6d073df82cbab6cce7a17ad4803bc072
|
|
| BLAKE2b-256 |
0756fa1c0e4cc281c4b4d7569b05b797aaf7ee0f9dbc3c155e3adde73149480b
|







