Picking Unique Relevant Peptides for viraL Experiments

Project description
╔═══╦╗░╔╦═══╦═══╦╗░░╔═══╗
║╔═╗║║░║║╔═╗║╔═╗║║░░║╔══╝
║╚═╝║║░║║╚═╝║╚═╝║║░░║╚══╗
║╔══╣║░║║╔╗╔╣╔══╣║░╔╣╔══╝
║║░░║╚═╝║║║╚╣║░░║╚═╝║╚══╗
╚╝░░╚═══╩╝╚═╩╝░░╚═══╩═══╝

Picking Unique Relevant Peptides for viraL Experiments
Version: 0.4.2
- Description
- Requirements
- Clone
- Target Selection
- How-To
- How-To-pip
- How-To-Conda
- Configuration
- Output
- Workflow
Description
Emerging virus diseases present a global threat to public health. To detect viral pathogens in time-critical scenarios, accurate and fast diagnostic assays are required. Such assays can now be established using mass spectrometry-based targeted proteomics, by which viral proteins can be rapidly detected from complex samples down to the strain level with high sensitivity and reproducibility. Developing such targeted assays involves tedious steps of peptide candidate selection, peptide synthesis, and assay optimization. Peptide selection requires extensive preprocessing by comparing candidate peptides against a large search space of background proteins. Here we present Purple (Picking unique relevant peptides for viral experiments), a software tool for selecting target-specific peptide candidates directly from given proteome sequence data. It comes with an intuitive graphical user interface, various parameter options and a threshold-based filtering strategy for homologous sequences. Purple enables peptide candidate selection across various taxonomic levels and filtering against backgrounds of varying complexity. Its functionality is demonstrated using data from different virus species and strains. Our software enables to build taxon-specific targeted assays and paves the way to time-efficient and robust viral diagnostics using targeted proteomics.
Requirements
- Python 3.4+
- tqdm
- biopython
- pyyaml
Clone
git clone https://gitlab.com/HartkopfF/Purple
Target Selection
Only the root directory is used and all subdirectories are excluded as well as all files not ending with the .fasta ending. Two options of target selection are implemented. The first one is to name targets in a list separated by a comma. Using this method, all databases are merged and every protein that is containing one of the targets in the origin species (OS) part of the UniProt header is considered as a target protein. The process of origin species matching is not case sensitive. Non-target proteins are used as background database. The second method is to specify one file in the database directory as target database. All remaining databases are merged and are assembled as background database. As the background database could still consist of proteins originating in one of the target species, every protein in the background database is removed from further analysis if it matches a target species in the target database.
How to use Purple
-
Download the latest version from the releases page (no Python required).
-
Double-clicking the downloaded executable file starts Purple.
-
Load a configuration file or edit parameters (database folder and target selection) via graphical user interface.
-
Open results in the output folder
How to use Purple directly in python via pip
Purple is available on PyPi here:
- Install the latest version with:
pip install purple-bio
or
pip3 install purple-bio
-
Edit the config file config.yml (download template) and specify database folder and target.
-
Add these lines to your python 3.x code:
import purple
purple.main("path/to/config.yml")
- Open results in the output folder
How to use Purple directly in Conda
- Install the latest version with:
conda install purple-bio
-
Edit the config file config.yml (download template) and specify database folder and target.
-
Add these lines to your python 3.x code:
import purple
purple.main("path/to/config.yml")
- Open results in the output folder
Configuration
Configuration yaml file template (download):
purple:
comment: comment
i_am_not_sure_about_target: true
leucine_distincion: false
leucine_distinction: false
max_len_peptides: 50
min_len_peptides: 5
path_DB: ../res/DB
path_output: ../output/
print_peptides: true
proline_digestion: false
removeFragments: true
target: [target1,target2]
targetFile: path/to/targetFile
threshold: 80
update_DB: true
List of parameters
| Parameter | Description | Example | Default |
|----------------------------|-------------------------------------------------------------|----------------------------|------------|
| target | List of targets to find unique peptides | [Hepatitis B, Hepatitis A] | No default |
| threshold | Threshold to filter matches | Values between 0 and 100 | 70 |
| update_DB | Build a database or use old one | True or False | False |
| path_DB | Path to folder with fasta files | C:/myFASTAs/ | ../res/DB/ |
| path_output | Path to output folder to store results | C:/results/ | ../output/ |
| targetFile | File name of the fasta with target entries | target.fasta | |
| i_am_not_sure_about_target | Option to check targets before matching peptides | True or False | True |
| max_len_peptides | Maximum length of peptides | Positive numerical values | 25 |
| min_len_peptides | Minimum length of peptides | Positive numerical values | 5 |
| removeFragments | Option to remove proteins with "(Fragments)" in the header | True or False | No default |
| leucine_distinction | Option to enable distinction of leucine and isoleucine | True or False | No default |
| proline_digestion | Option to apply proline digestion rule | True or False | No default |
| print_peptides | Print peptides at the end | True or False | False |
| comment | Comments for the log book | Text or numbers | no comment |
Output
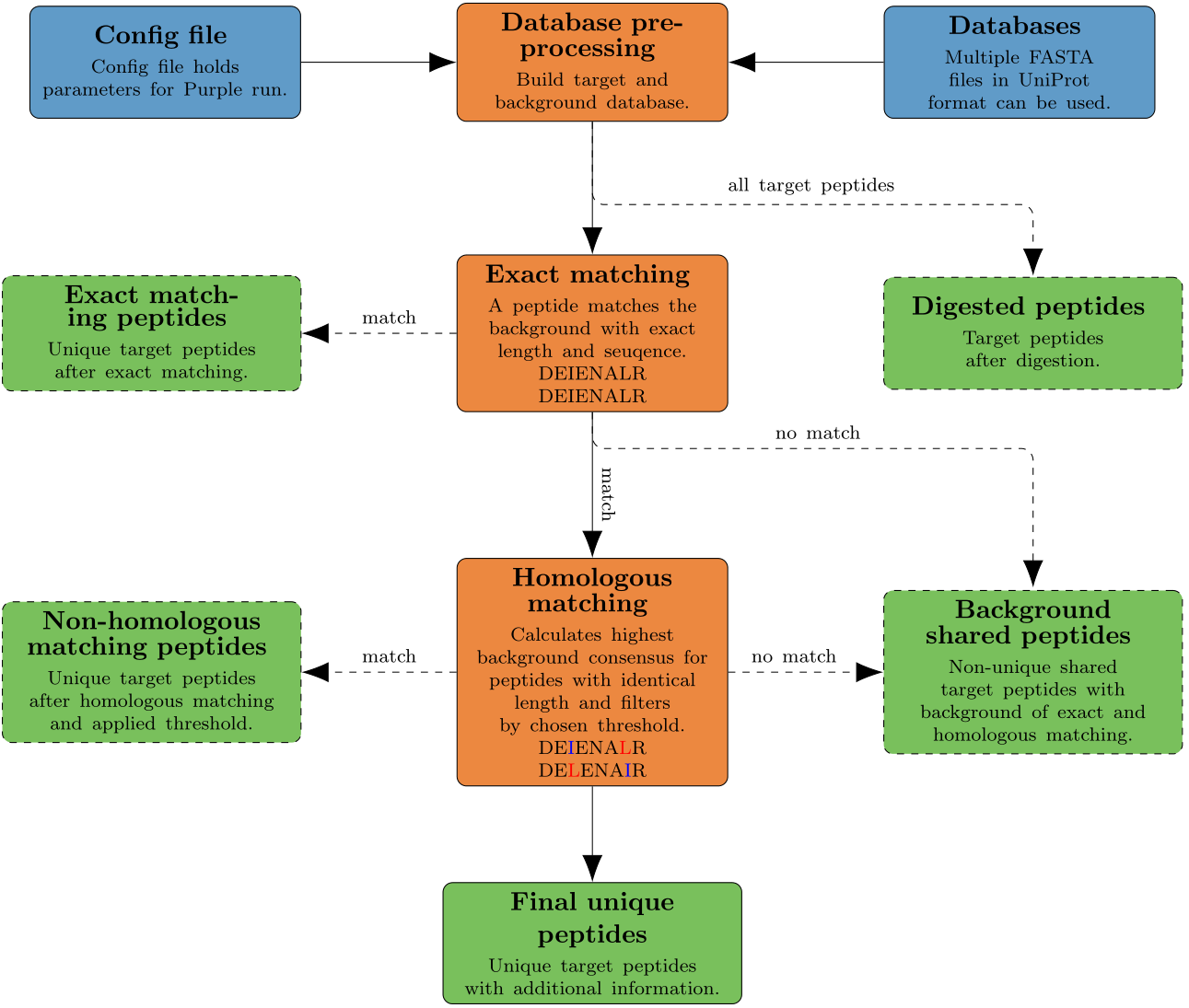
The output includes a folder with seven files for shared, exact matching, homologous matching, digested and final unique peptides for a specifc target. Additionally, a log of the command line output of Purple and a logbook with a short summary of the run is provided.
Information in final results file:
- Peptide: Unique peptide sequence.
- Peptide weight: Peptide weight of the unique peptide calculated with Biopython.
- Highest background consensus: Highest background consensus of the homologous matching for each peptide.
- Occurrences: Number of occurrences for each peptide.
- Species: Species of the peptide.
- Protein name: Names of the proteins containing this peptide.
- Fasta entries: Headers of the fasta entries containing this peptide.
- Description: Complete header of the proteins listed in protein name.
Workflow


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file purple_bio-0.4.2.5.tar.gz.
File metadata
- Download URL: purple_bio-0.4.2.5.tar.gz
- Upload date:
- Size: 16.1 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/1.13.0 pkginfo/1.5.0.1 requests/2.21.0 setuptools/41.0.0 requests-toolbelt/0.9.1 tqdm/4.32.2 CPython/3.7.3
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
3513a72c99352f8749aa689f4b8a6e3ce218029a9d2c2a63308e47ee9a146089
|
|
| MD5 |
3ec1cd89d605b53faa12f3b1422957ef
|
|
| BLAKE2b-256 |
f3e94bd8acdb60f090f78fc327cdce5ff0454ec62f09b8b51fae13ebe905f5e2
|
File details
Details for the file purple_bio-0.4.2.5-py3-none-any.whl.
File metadata
- Download URL: purple_bio-0.4.2.5-py3-none-any.whl
- Upload date:
- Size: 16.2 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/1.13.0 pkginfo/1.5.0.1 requests/2.21.0 setuptools/41.0.0 requests-toolbelt/0.9.1 tqdm/4.32.2 CPython/3.7.3
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
96601e92c384f10ed7531c315223127e96143450a093447e31ebff6e77e2a476
|
|
| MD5 |
04e4a743f924400de1b3561bfe7de2db
|
|
| BLAKE2b-256 |
ce885e95bbb785ea47a6cab14275d96a6dded19c59bfdeffa3bd832f44c31271
|







