XANES quantification of functional group abundances


Project description
QUANTORXS is an open-source program to automatically analyze XANES spectra at Carbon, Nitrogen and Oxygen K-edges edges to quantify the concentration of functional groups and the elemental ratios (N/C and O/C). It is based on a novel quantification method published in Analytical chemistry.
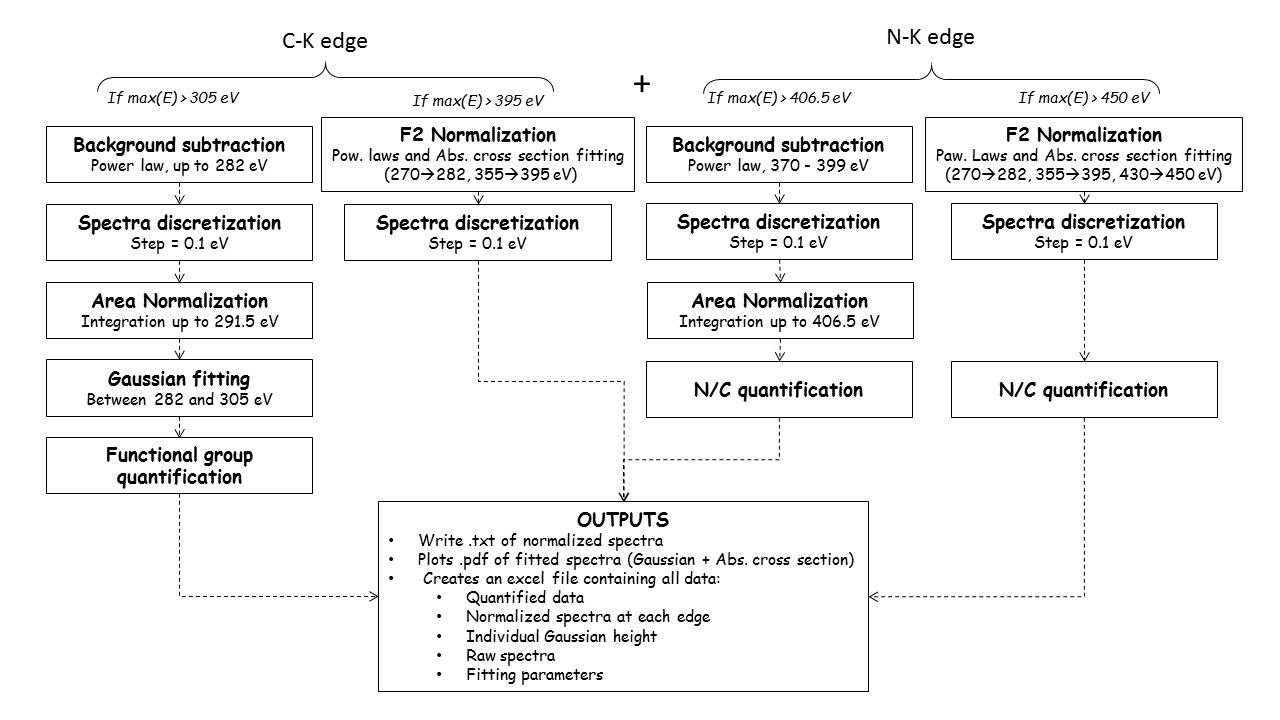
QUANTORXS performs the following tasks automatically:
Load the data from the file(s)
Remove background
Normalize the spectra
Generate a model of the fine structure a fit it to the experimental data
Calculate the functional groups abundances and elemental rations from the results of the fit
Generate an Excel file and multiple figures with the results and normalised spectra files.
This is illustrated in more detail in the following diagram:
Alt text
QUANTORXS is designed to work without any user input other than the experimental spectra. Users willing to modify the details of the quantification can download the code from its GitHub repository.
The code was initially written by Corentin Le Guillou. Francisco de la Peña created the command line and graphical user interfaces.
Installing QUANTORXS.
QUANTORXS is written in the Python programming languague and is available from pypi. It runs in any operating system with the Python programming language installed.
To install QUANTORXS execute the following in a terminal:
pip install quantorxsStep-by-step installation instructions for Windows users
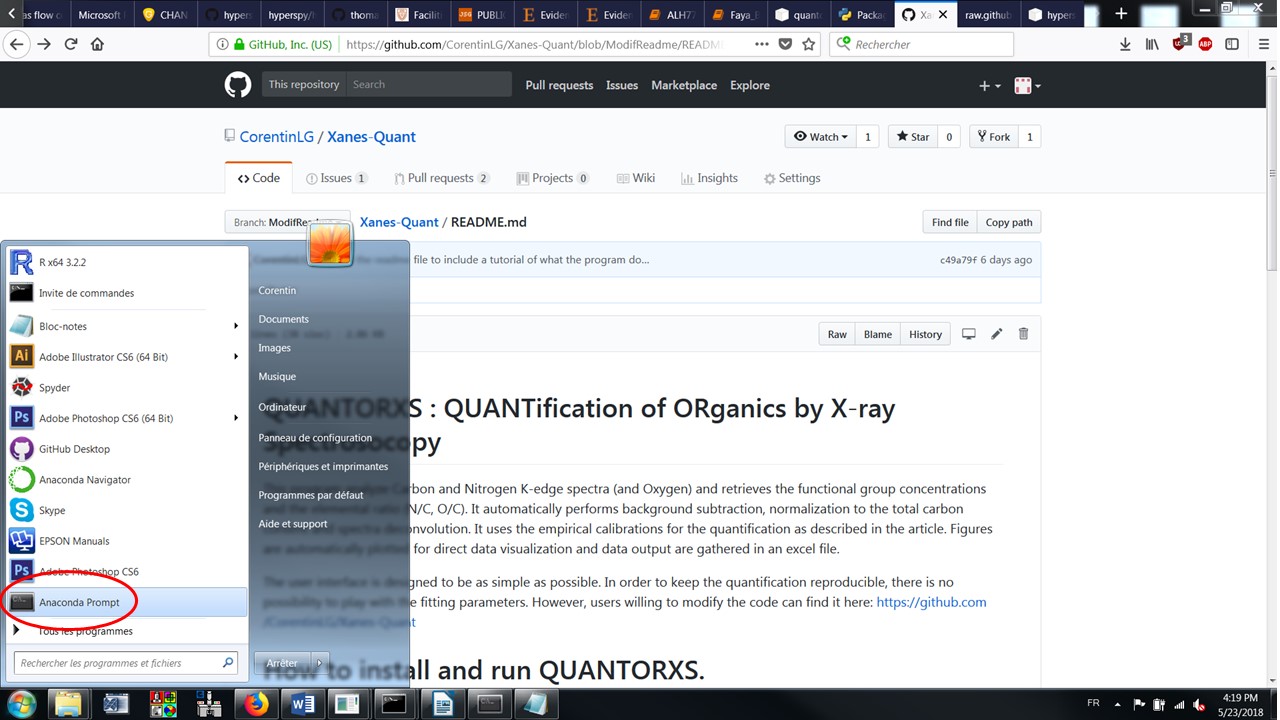
If you are new to Python we reccomend you to install the opensource and free Anaconda Python distribution for your platform first. Afterwards, from the Microsoft Windows Start Menu, open “anaconda prompt” as in the image below:
Alt text
Then type the following and press Enter (requires connection to the internet):
pip install quantorxs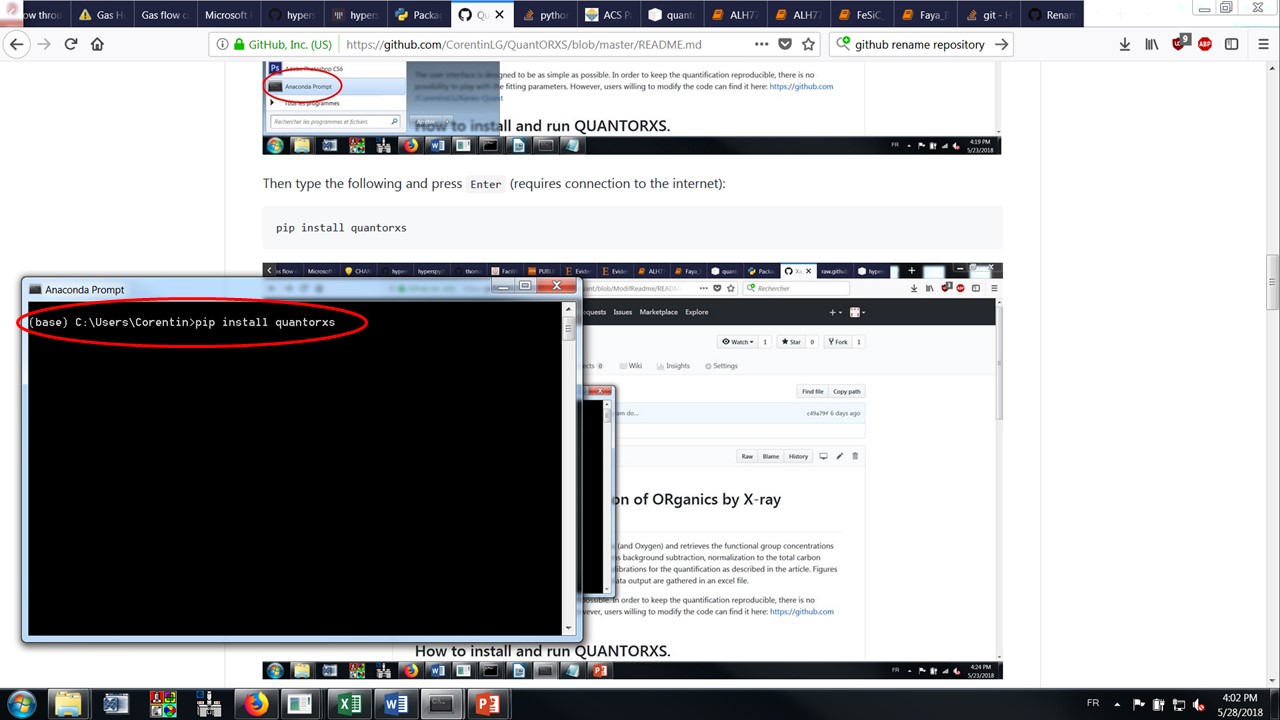
Alt text
That’s all! QUANTORXS should now be installed in your system.
Starting the QUANTORXS Graphical User Interface
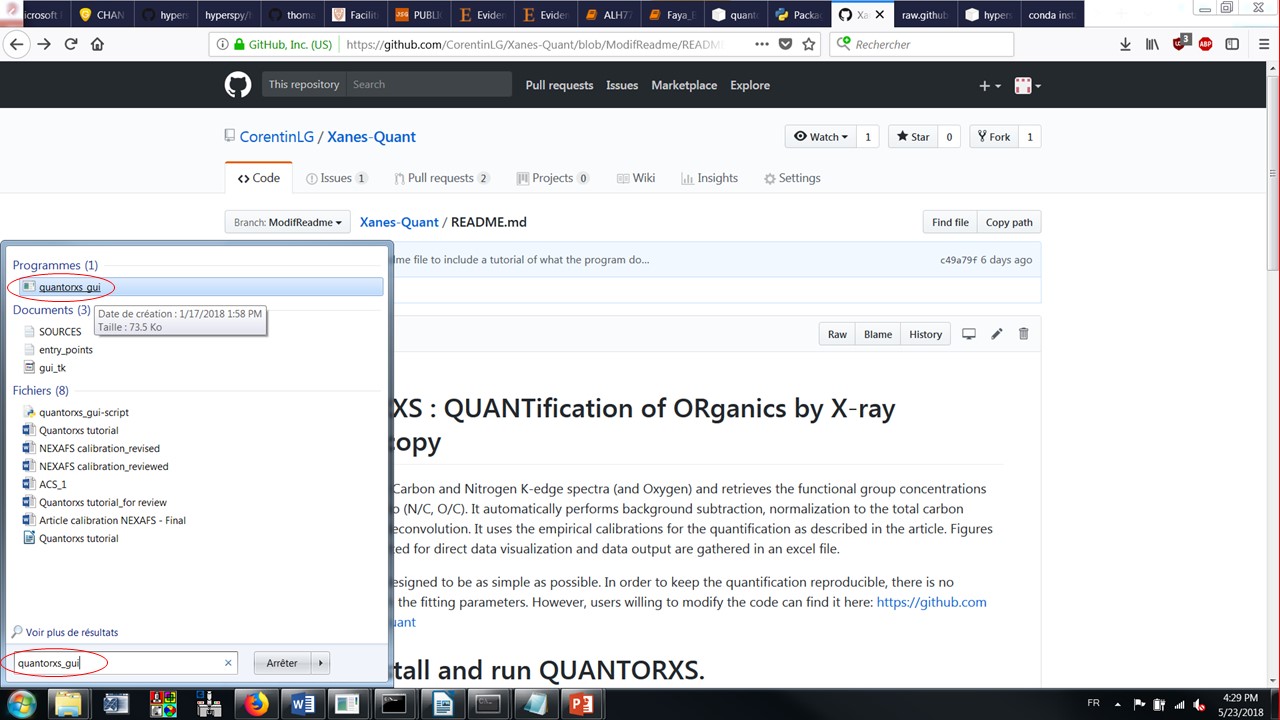
To start the graphical interface execute the quantorxs_gui e.g. a terminal. Alternatively, Windows users can start it by searching for the executable file “quantorxs_gui” in the Start Menu and launching it as shown in the image below.
Alt text
How to use the graphical interface
The program is designed to process several spectra at once. All source spectra should be assembled in one folder. QUANTORXS reads only the format produced by aXis2000
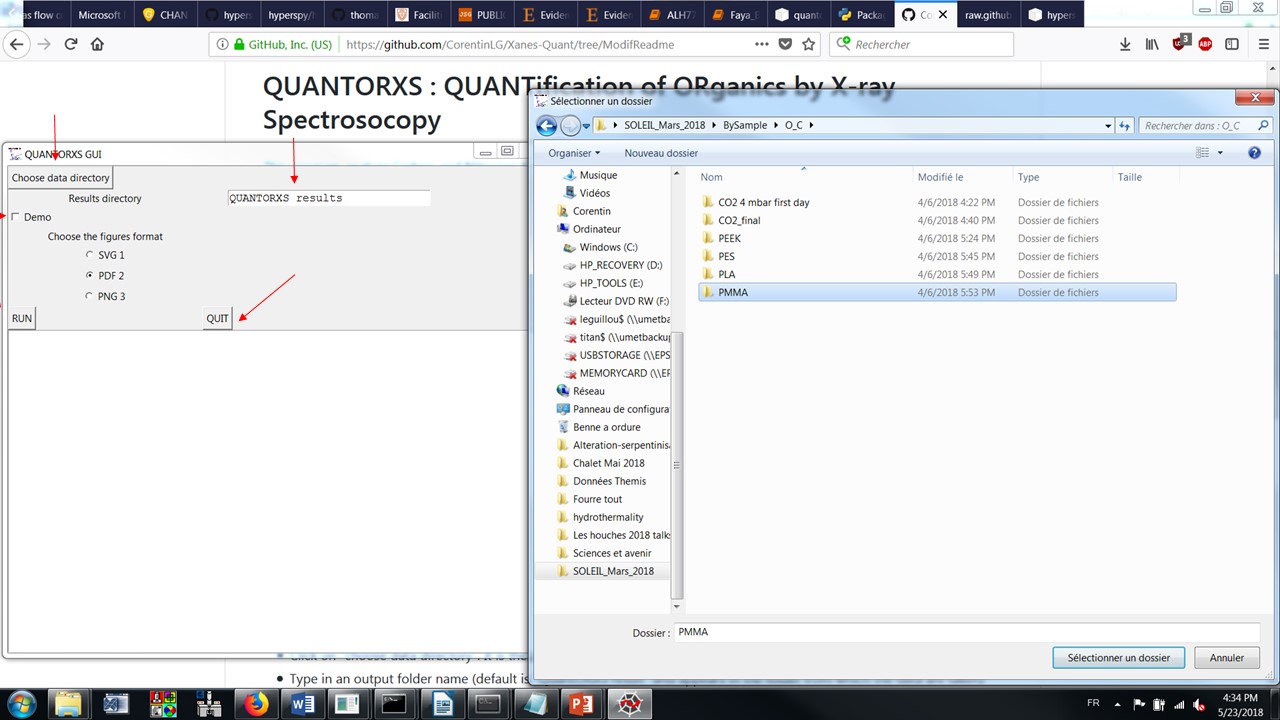
Click on the Choose data directory button and select the folder containing the source spectra.
Type in an output folder name (relative to the data directory) to store the results of the analysis. The default is QUANTORXS results.
Make sure that the demo box is not checked. If checked, it uses default files as input to produce an example of the output files.
Select the format of the figure output (the default is SVG)
Set the offset if required to compensate from any energy misalignment (e.g. from poorly calibrated monochromator) common to all spectra.
Click the Run button and wait until the analysis is completed (usually a few secondes per spectrum).
Alt text
Description of the output files
The output folder will be created in the folder from which the data have been taken. An .xls result file and two different sub-folders are created:
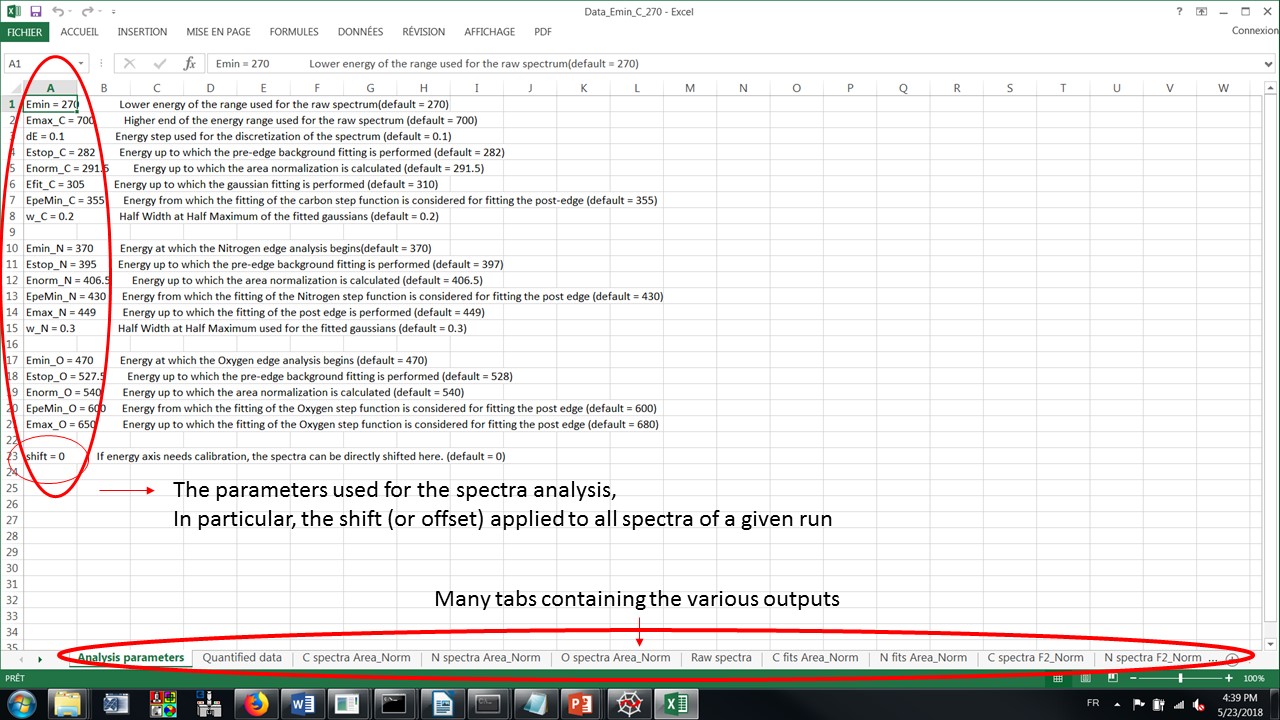
a .xls file contains several sheets:
The fitting parameters
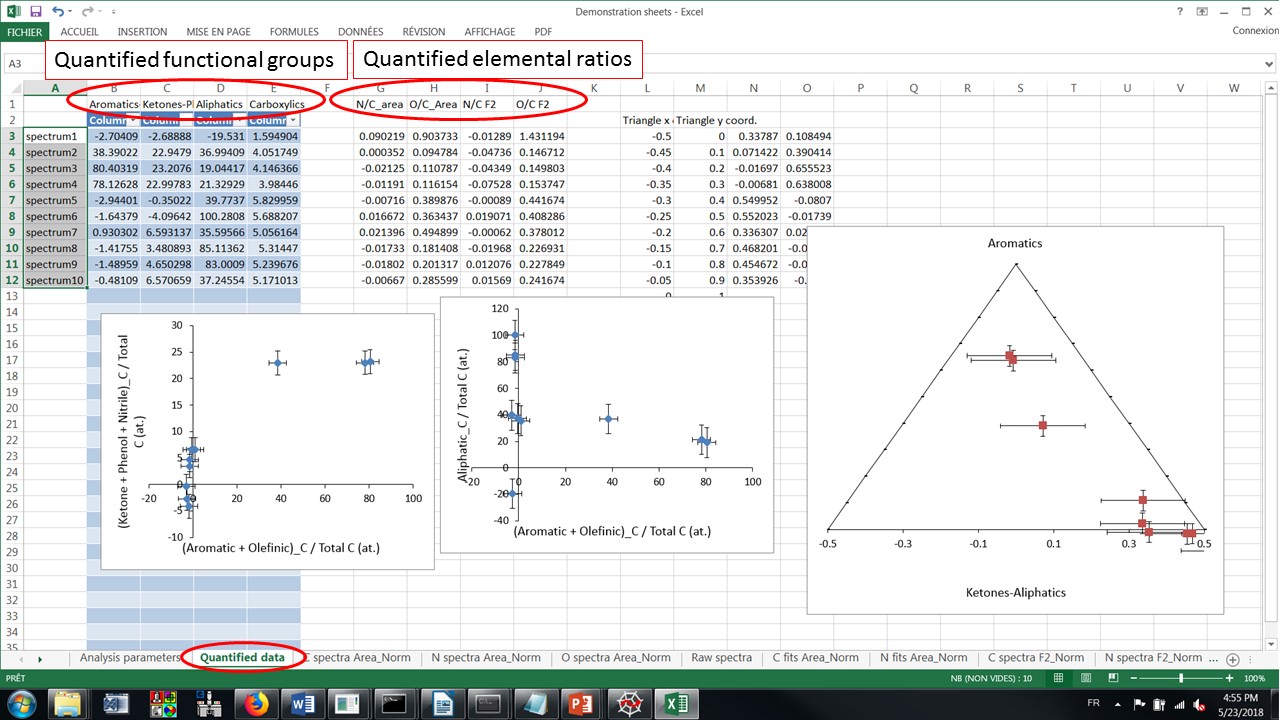
The quantified data (aromatic, ketones, aliphatics, carboxylics; as well as N/C and O/C ratios) and some related plots
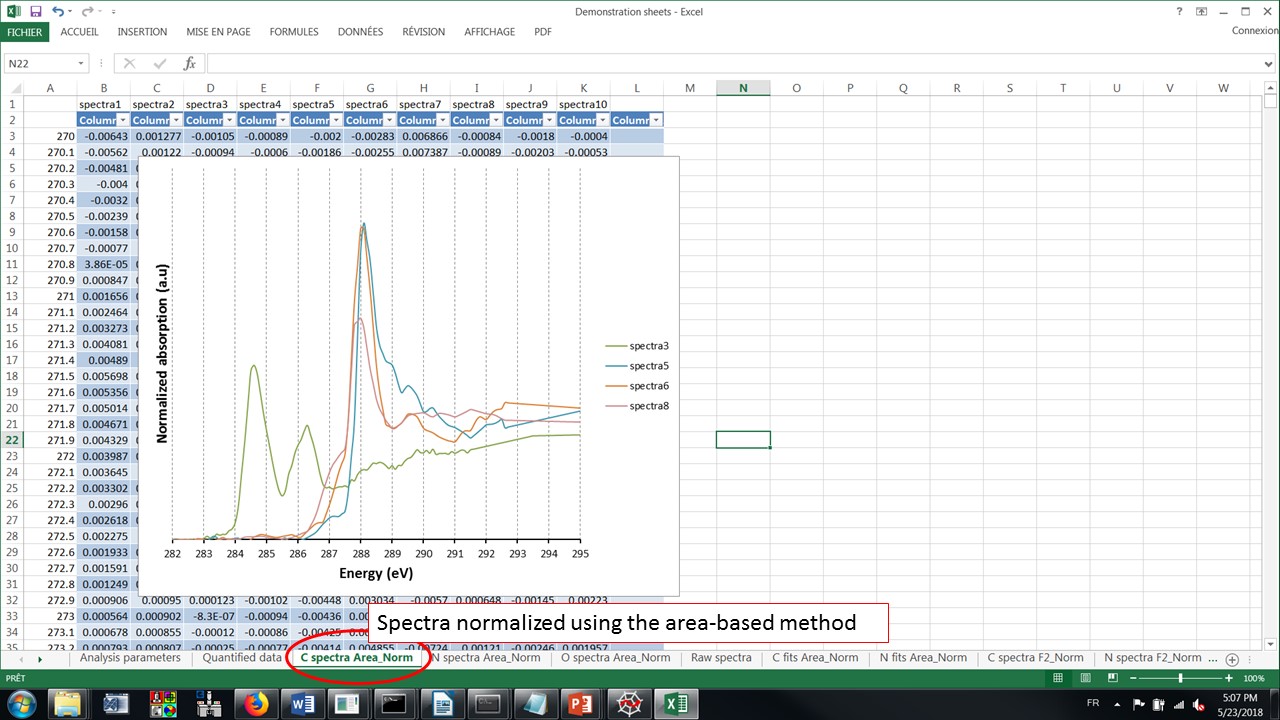
The spectra at the C-K edge normalized by the area ratio method
The spectra at the N-K edge normalized by the area ratio method
The spectra at the O-K edge normalized by the area ratio method
The fitted heights of the Gaussians for the area-based normalization at the C-K edge
The fitted heights of the Gaussians for the area-based normalization at the N-K edge
The fitted heights of the Gaussians for the area-based normalization at the O-K edge
Alt text
Alt text
Alt text
Alt text
A folder containing the .txt files of each normalized spectrum
A folder with figures displaying:
The cross-section fit
The normalized spectra
The deconvolution (all gaussians included)
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file quantorxs-0.1.1.tar.gz.
File metadata
- Download URL: quantorxs-0.1.1.tar.gz
- Upload date:
- Size: 78.2 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
b6164c563d3b926c3351cc03b41f2a6c418329d1ea7f2807c5d917de3bca9032
|
|
| MD5 |
990902ae6c2e23fb1573c1ec2109eac7
|
|
| BLAKE2b-256 |
65d46b764ed5fbd8fe319cb6a3f654d4a87ed6e01c625181b8ab1f2775d6151e
|







