


Qupid
(Pronounced like cupid)
Qupid is a tool for generating and statistically evaluating multiple case-control matchings of microbiome data.
Installation
You can install the most up-to-date version of Qupid from PyPi using the following command:
pip install qupid
Quickstart
Qupid provides a convenience function, shuffle, to easily generate multiple matches based on matching critiera.
This block of code will determine each viable control per case and randomly pick 10 arrangments of a single case matched to a single valid control.
The output is a pandas DataFrame where the rows are case names and each column represents a valid mapping of case to control.
from pkg_resources import resource_filename
import pandas as pd
import qupid
metadata_fpath = resource_filename("qupid", "tests/data/asd.tsv")
metadata = pd.read_table(metadata_fpath, sep="\t", index_col=0)
asd_str = "Diagnosed by a medical professional (doctor, physician assistant)"
no_asd_str = "I do not have this condition"
background = metadata.query("asd == @no_asd_str")
focus = metadata.query("asd == @asd_str")
matches = qupid.shuffle(
focus=focus,
background=background,
categories=["sex", "age_years"],
tolerance_map={"age_years": 10},
iterations=100
)
Tutorial
There are three primary steps to the Qupid workflow:
- Match each case to all valid controls
- Generate multiple one-to-one matchings
- Evaluate the statistical differences between cases and controls for all matchings
To match each case to all valid controls, we need to first establish matching criteria. Qupid allows matching by both categorical metadata (exact matches) and continuous metadata (matching within provided tolerance). You can match on either a single metadata column or based on multiple.
In Qupid, the cases to be matched are referred to as the "focus" set, while the set of all possible controls is called the "background". For this tutorial we will be used data from the American Gut Project to match cases to controls in samples from people with autism.
First, we'll load in the provided example metadata and separate it into the focus (samples from people with autism) and the background (samples from people who do not have autism).
Loading data
from pkg_resources import resource_filename
import pandas as pd
metadata_fpath = resource_filename("qupid", "tests/data/asd.tsv")
metadata = pd.read_table(metadata_fpath, sep="\t", index_col=0)
# Designate focus samples
asd_str = "Diagnosed by a medical professional (doctor, physician assistant)"
no_asd_str = "I do not have this condition"
background = metadata.query("asd == @no_asd_str")
focus = metadata.query("asd == @asd_str")
Matching each case to all possible controls
Next, we want to perform case-control matching on sex and age.
Sex is a discrete factor, so Qupid will attempt to find exact matches (e.g. male to male, female to female).
However, age is a continuous factor; as a result, we should provide a tolerance value (e.g. match within 10 years).
We use the match_by_multiple function to match based on more than one metadata category.
from qupid import match_by_multiple
cm = match_by_multiple(
focus=focus,
background=background,
categories=["sex", "age_years"],
tolerance_map={"age_years": 10}
)
This creates a CaseMatchOneToMany object where each case is matched to each possible control.
You can view the underlying matches as a dictionary with cm.case_control_map.
Generating mappings from each case to a single control
What we now want is to match each case to a single control so we can perform downstream analysis. However, we have a lot of possible controls. We can easily see how many cases and possible controls we have.
print(len(cm.cases), len(cm.controls))
This tells us that we have 45 cases and 1785 possible controls. Because of this, there are many possible sets of valid matchings of each case to a single control. We can use Qupid to generate many such cases.
results = cm.create_matched_pairs(iterations=100)
This creates a CaseMatchCollection data structure that contains 100 CaseMatchOneToOne instances.
Each CaseMatchOneToOne entry maps each case to a single control rather than all possible controls.
We can verify that each entry has exactly 45 cases and 45 controls.
print(len(results[0].cases), len(results[0].controls))
Qupid provides a convenience method to convert a CaseMatchCollection object into a pandas DataFrame.
The DataFrame index corresponds to the cases, while each column represents a distinct set of matching controls.
The value in a cell represents a matching control to the row's case.
results_df = results.to_dataframe()
results_df.head()
0 1 ... 98 99
case_id ...
S10317.000026181 S10317.000033804 S10317.000069086 ... S10317.000108605 S10317.000076381
S10317.000071491 S10317.000155409 S10317.000103912 ... S10317.000099277 S10317.000036401
S10317.000029293 S10317.000069676 S10317.X00175749 ... S10317.000069299 S10317.000066846
S10317.000067638 S10317.X00179103 S10317.000052409 ... S10317.000067511 S10317.000067601
S10317.000067637 S10317.000067747 S10317.000098161 ... S10317.000017116 S10317.000067997
[5 rows x 100 columns]
Statistical assessment of matchings
Once we have this list of matchings, we want to determine how statistically difference cases are from controls based on some values. Qupid supports two types of statistical tests: univariate and multivariate. Univariate data is in the form of a vector where each case and control has a single value. This can be alpha diversity, log-ratios, etc. Multivariate data is in the form of a distance matrix where each entry is the pairwise distance between two samples, e.g. from beta diversity analysis. We will generate random data for this tutorial where there exists a small difference between ASD samples and non-ASD samples.
import numpy as np
rng = np.random.default_rng()
asd_mean = 4
ctrl_mean = 3.75
num_cases = len(cm.cases)
num_ctrls = len(cm.controls)
asd_values = rng.normal(asd_mean, 1, size=num_cases)
ctrl_values = rng.normal(ctrl_mean, 1, size=num_ctrls)
asd_values = pd.Series(asd_values, index=focus.index)
ctrl_values = pd.Series(ctrl_values, index=background.index)
sample_values = pd.concat([asd_values, ctrl_values])
We can now evaluate a t-test between case values and control values for each possible case-control matching in our collection.
from qupid.stats import bulk_univariate_test
test_results = bulk_univariate_test(
casematches=results,
values=sample_values,
test="t"
)
This returns a DataFrame of test results sorted by descending test statistic.
method_name test_statistic_name test_statistic p-value sample_size number_of_groups
15 t-test t 3.900874 0.000187 90 2
61 t-test t 3.770914 0.000294 90 2
50 t-test t 3.536803 0.000649 90 2
32 t-test t 3.395298 0.001030 90 2
68 t-test t 3.310822 0.001350 90 2
.. ... ... ... ... ... ...
13 t-test t 0.645694 0.520158 90 2
49 t-test t 0.555063 0.580260 90 2
92 t-test t 0.409252 0.683349 90 2
51 t-test t 0.110707 0.912101 90 2
34 t-test t 0.048571 0.961371 90 2
[100 rows x 6 columns]
From this table, we can see that iteration 15 best separates cases from controls based on our random data. Conversely, iteration 34 showed essentially no difference between cases and controls. This shows that it is important to create multiple matchings as some of them are better than others. We can plot the distribution of p-values to get a sense of the overall distribution.
import matplotlib.pyplot as plt
import seaborn as sns
sns.histplot(test_results["p-value"])
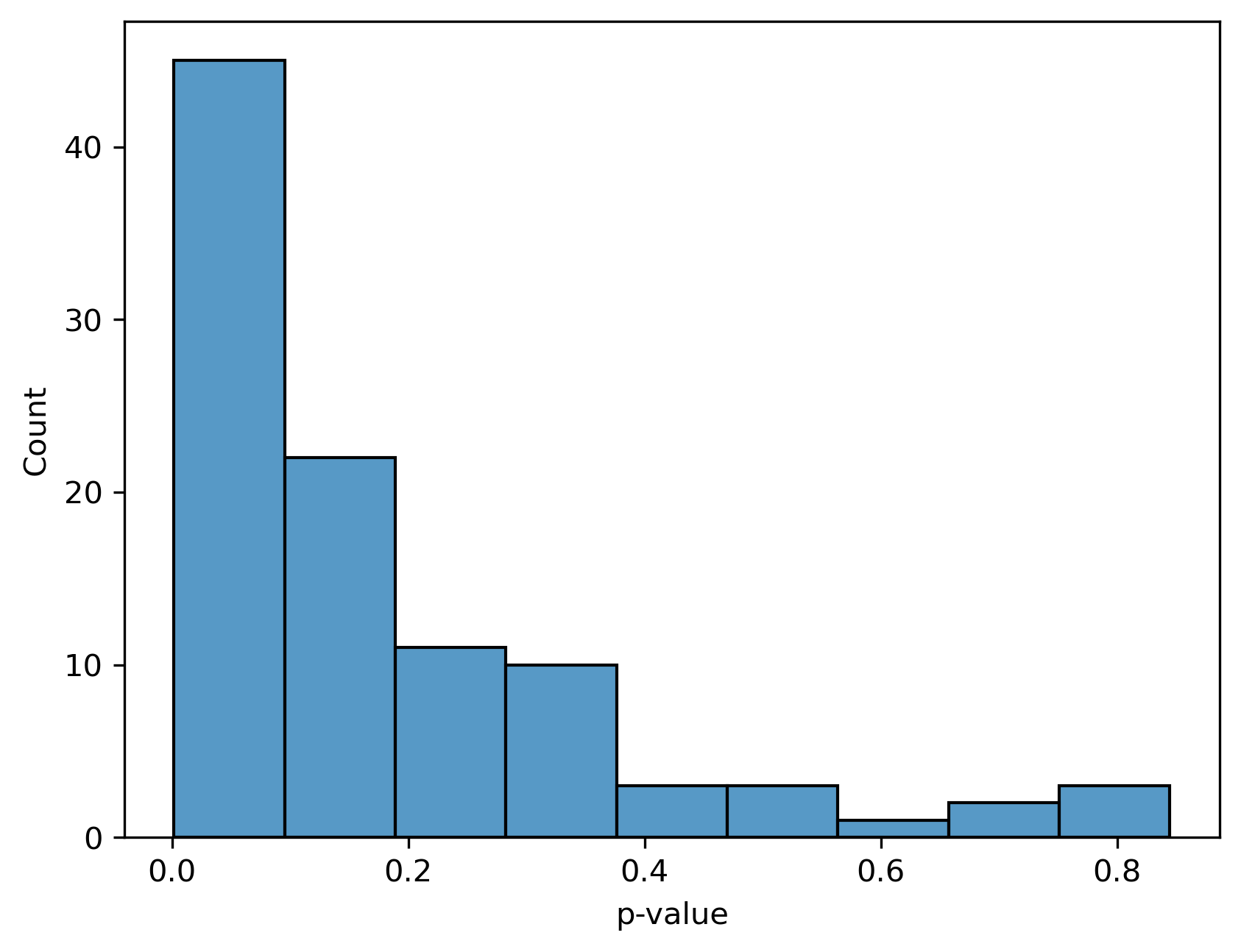
We see that most of the p-values are near zero which makes sense because we simulated our data with a difference between ASD and non-ASD samples.
Saving and loading qupid results
Qupid allows the saving and loading of both CaseMatch and CaseMatchCollection objects.
CaseMatchOneToMany and CaseMatchOneToOne objects are saved as JSON files while CaseMatchCollection objects are saved as pandas DataFrames.
from qupid.casematch import CaseMatchOneToMany, CaseMatchOneToOne, CaseMatchCollection
cm.save("asd_matches.one_to_many.json") # Save all possible matches
results.save("asd_matches.100.tsv") # Save all 100 iterations
results[15].save("asd_matches.best.json") # Save best matching
CaseMatchOneToMany.load("asd_matches.one_to_many.json")
CaseMatchCollection.load("asd_matches.100.tsv")
CaseMatchOneToOne.load("asd_matches.best.json")
Command Line Interface
Qupid has a command line interface to create multiple matchings from cases and possible controls.
If providing numeric categories, the column name must be accompanied by the tolerance after a space (e.g. age_years 5 for a tolerance of 5 years).
You can pass multiple options to --discrete-cat or --numeric-cat to specify multiple matching criteria.
For usage detalls, use qupid shuffle --help.
qupid shuffle \
--focus focus.tsv \
--background background.tsv \
--iterations 15 \
--discrete-cat sex \
--discrete-cat race \
--numeric-cat age_years 5 \
--numeric-cat weight_lbs 10 \
--output matches.tsv
QIIME 2 Usage
Qupid provides support for the popular QIIME 2 framework of microbiome data analysis. We assume in this tutorial that you are familiar with using QIIME 2 on the command line. If not, we recommend you read the excellent documentation before you get started with Qupid.
Run qiime qupid --help to see all possible commands.
Matching one-to-many
Use qiime qupid match-one-to-many to match each case to all possible controls.
Note that for numeric categories, you must pass in tolerances in the form of <column_name>+-<tolerance>.
qiime qupid match-one-to-many \
--m-sample-metadata-file metadata.tsv \
--p-case-control-column case_control \
--p-categories sex age_years \
--p-case-identifier case \
--p-tolerances age_years+-10 \
--o-case-match-one-to-many cm_one_to_many.qza
Matching one-to-one
With a one-to-many match, you can generate multiple possible one-to-one matches using qiime qupid match-one-to-one.
qiime qupid match-one-to-one \
--i-case-match-one-to-many cm_one_to_many.qza \
--p-iterations 10 \
--o-case-match-collection cm_collection.qza
Qupid shuffle
The previous two commands can be run sequentially using qiime qupid shuffle.
qiime qupid shuffle \
--m-sample-metadata-file metadata.tsv \
--p-case-control-column case_control \
--p-categories sex age_years \
--p-case-identifier case \
--p-tolerances age_years+-10 \
--p-iterations 10 \
--output-dir shuffle
Statistical assessment of matches
You can assess how different cases are from controls using both univariate data (such as alpha diversity) or multivariate data (distance matrices). The result will be a histogram of p-values from either a t-test (univariate) or PERMANOVA (multivariate) comparing cases to controls. Note that for either command, the input data must contain values for all possible cases and controls.
qiime qupid assess-matches-univariate \
--i-case-match-collection cm_collection.qza \
--m-data-file data.tsv \
--m-data-column faith_pd \
--o-visualization univariate_p_values.qzv
qiime qupid assess-matches-multivariate \
--i-case-match-collection cm_collection.qza \
--i-distance-matrix uw_unifrac_distance_matrix.qza \
--p-permutations 999 \
--o-visualization multivariate_p_values.qzv
Help with Qupid
If you encounter a bug in Qupid, please post a GitHub issue and we will get to it as soon as we can. We welcome any ideas or documentation updates/fixes so please submit an issue and/or a pull request if you have thoughts on making Qupid better.
1 maintainer
Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file qupid-0.1.0.tar.gz.
File metadata
- Download URL: qupid-0.1.0.tar.gz
- Upload date:
- Size: 41.4 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/0.0.0 CPython/3.8.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
32d7dc339105656f92198ca5ce4990deac332bed980e00f5d13677676a73474d
|
|
| MD5 |
72b21173b1fa4801da93b702872e2ff0
|
|
| BLAKE2b-256 |
c1fdde99567b7653fb61a6ca352e65c403b26ec213ed22fbb06c6bb518db29e4
|









