Rule Set 3
Python package to predict the activity of CRISPR sgRNA sequences using Rule Set 3
Install
You can install the latest release of rs3 from pypi using
pip install rs3
For mac users you may also have to brew install the OpenMP library
brew install libomp
or install lightgbm without Openmp
pip install lightgbm --install-option=--nomp
See the LightGBM documentation for more information
Documentation
You can see the complete documentation for Rule Set 3 here.
Quick Start
Sequence based model
To calculate Rule Set 3 (sequence) scores, import the predict_seq function from the seq module.
from rs3.seq import predict_seq
You can store the 30mer context sequences you want to predict as a list.
context_seqs = ['GACGAAAGCGACAACGCGTTCATCCGGGCA', 'AGAAAACACTAGCATCCCCACCCGCGGACT']
You can specify the Hsu2013 or Chen2013 as the tracrRNA to score with. We generally find any tracrRNA that does not have a T in the fifth position is better predicted with the Chen2013 input.
predict_seq(context_seqs, sequence_tracr='Hsu2013')
Calculating sequence-based features
100%|██████████| 2/2 [00:00<00:00, 352.28it/s]
array([-0.90030944, 1.11451622])
Target based model
To get target scores, which use features at the endogenous target site to make predictions, you must build or load feature matrices for the amino acid sequences, conservation scores, and protein domains.
As an example, we'll calculate target scores for 250 sgRNAs in the GeckoV2 library.
import pandas as pd
from rs3.predicttarg import predict_target
from rs3.targetfeat import (add_target_columns,
get_aa_subseq_df,
get_protein_domain_features,
get_conservation_features)
design_df = pd.read_table('test_data/sgrna-designs.txt')
design_df.head()
.dataframe tbody tr th {
vertical-align: top;
}
.dataframe thead th {
text-align: right;
}
| Input | Quota | Target Taxon | Target Gene ID | Target Gene Symbol | Target Transcript | Target Reference Coords | Target Alias | CRISPR Mechanism | Target Domain | ... | On-Target Rank Weight | Off-Target Rank Weight | Combined Rank | Preselected As | Matching Active Arrayed Oligos | Matching Arrayed Constructs | Pools Containing Matching Construct | Pick Order | Picking Round | Picking Notes | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | PSMB7 | 2 | 9606 | ENSG00000136930 | PSMB7 | ENST00000259457.8 | NaN | NaN | CRISPRko | CDS | ... | 1.0 | 1.0 | 7 | GCAGATACAAGAGCAACTGA | NaN | BRDN0004619103 | NaN | 1 | 0 | Preselected |
| 1 | PSMB7 | 2 | 9606 | ENSG00000136930 | PSMB7 | ENST00000259457.8 | NaN | NaN | CRISPRko | CDS | ... | 1.0 | 1.0 | 48 | AAAACTGGCACGACCATCGC | NaN | NaN | NaN | 2 | 0 | Preselected |
| 2 | PRC1 | 2 | 9606 | ENSG00000198901 | PRC1 | ENST00000394249.8 | NaN | NaN | CRISPRko | CDS | ... | 1.0 | 1.0 | 7 | AAAAGATTTGCGCACCCAAG | NaN | NaN | NaN | 1 | 0 | Preselected |
| 3 | PRC1 | 2 | 9606 | ENSG00000198901 | PRC1 | ENST00000394249.8 | NaN | NaN | CRISPRko | CDS | ... | 1.0 | 1.0 | 8 | CTTTGACCCAGACATAATGG | NaN | NaN | NaN | 2 | 0 | Preselected |
| 4 | TOP1 | 2 | 9606 | ENSG00000198900 | TOP1 | ENST00000361337.3 | NaN | NaN | CRISPRko | CDS | ... | 1.0 | 1.0 | 1 | NaN | NaN | BRDN0001486452 | NaN | 2 | 1 | NaN |
5 rows × 60 columns
Throughout the analysis we will be using a core set of ID columns to merge the feature matrices. These ID columns should uniquely identify an sgRNA and its target site.
id_cols = ['sgRNA Context Sequence', 'Target Cut Length', 'Target Transcript', 'Orientation']
Amino acid sequence input
To calculate the amino acid sequence matrix, you must first load the complete sequence from ensembl using the
build_transcript_aa_seq_df. See the documentation for the predicttarg module for an example of how to
use this function.
In this example we will use amino acid sequences that have been precalculated using the write_transcript_data
function in the targetdata module. Check out the documentation for this module for more information on
how to use this function.
We use pyarrow to read the written transcript data.
The stored transcripts are indexed by their Ensembl ID without the version number identifier.
To get this shortened version of the Ensembl ID use the add_target_columns function from the targetfeat module.
This function adds the 'Transcript Base' column as well as a column indicating the amino acid index ('AA Index')
of the cut site. The 'AA Index' column will be used for merging with the amino acid translations.
design_targ_df = add_target_columns(design_df)
design_targ_df.head()
.dataframe tbody tr th {
vertical-align: top;
}
.dataframe thead th {
text-align: right;
}
| Input | Quota | Target Taxon | Target Gene ID | Target Gene Symbol | Target Transcript | Target Reference Coords | Target Alias | CRISPR Mechanism | Target Domain | ... | Combined Rank | Preselected As | Matching Active Arrayed Oligos | Matching Arrayed Constructs | Pools Containing Matching Construct | Pick Order | Picking Round | Picking Notes | AA Index | Transcript Base | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | PSMB7 | 2 | 9606 | ENSG00000136930 | PSMB7 | ENST00000259457.8 | NaN | NaN | CRISPRko | CDS | ... | 7 | GCAGATACAAGAGCAACTGA | NaN | BRDN0004619103 | NaN | 1 | 0 | Preselected | 64 | ENST00000259457 |
| 1 | PSMB7 | 2 | 9606 | ENSG00000136930 | PSMB7 | ENST00000259457.8 | NaN | NaN | CRISPRko | CDS | ... | 48 | AAAACTGGCACGACCATCGC | NaN | NaN | NaN | 2 | 0 | Preselected | 46 | ENST00000259457 |
| 2 | PRC1 | 2 | 9606 | ENSG00000198901 | PRC1 | ENST00000394249.8 | NaN | NaN | CRISPRko | CDS | ... | 7 | AAAAGATTTGCGCACCCAAG | NaN | NaN | NaN | 1 | 0 | Preselected | 106 | ENST00000394249 |
| 3 | PRC1 | 2 | 9606 | ENSG00000198901 | PRC1 | ENST00000394249.8 | NaN | NaN | CRISPRko | CDS | ... | 8 | CTTTGACCCAGACATAATGG | NaN | NaN | NaN | 2 | 0 | Preselected | 263 | ENST00000394249 |
| 4 | TOP1 | 2 | 9606 | ENSG00000198900 | TOP1 | ENST00000361337.3 | NaN | NaN | CRISPRko | CDS | ... | 1 | NaN | NaN | BRDN0001486452 | NaN | 2 | 1 | NaN | 140 | ENST00000361337 |
5 rows × 62 columns
transcript_bases = design_targ_df['Transcript Base'].unique()
transcript_bases[0:5]
array(['ENST00000259457', 'ENST00000394249', 'ENST00000361337',
'ENST00000368328', 'ENST00000610426'], dtype=object)
aa_seq_df = pd.read_parquet('test_data/target_data/aa_seqs.pq', engine='pyarrow',
filters=[[('Transcript Base', 'in', transcript_bases)]])
aa_seq_df.head()
.dataframe tbody tr th {
vertical-align: top;
}
.dataframe thead th {
text-align: right;
}
| Target Transcript | Target Total Length | Transcript Base | version | seq | molecule | desc | id | AA len | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | ENST00000259457.8 | 834 | ENST00000259457 | 3 | MAAVSVYAPPVGGFSFDNCRRNAVLEADFAKRGYKLPKVRKTGTTI... | protein | None | ENSP00000259457 | 277 |
| 1 | ENST00000394249.8 | 1863 | ENST00000394249 | 3 | MRRSEVLAEESIVCLQKALNHLREIWELIGIPEDQRLQRTEVVKKH... | protein | None | ENSP00000377793 | 620 |
| 2 | ENST00000361337.3 | 2298 | ENST00000361337 | 2 | MSGDHLHNDSQIEADFRLNDSHKHKDKHKDREHRHKEHKKEKDREK... | protein | None | ENSP00000354522 | 765 |
| 3 | ENST00000368328.5 | 267 | ENST00000368328 | 4 | MALSTIVSQRKQIKRKAPRGFLKRVFKRKKPQLRLEKSGDLLVHLN... | protein | None | ENSP00000357311 | 88 |
| 4 | ENST00000610426.5 | 783 | ENST00000610426 | 1 | MPQNEYIELHRKRYGYRLDYHEKKRKKESREAHERSKKAKKMIGLK... | protein | None | ENSP00000483484 | 260 |
From the complete transcript translations, we extract an amino acid subsequence as input to our model. The subsequence is centered around the amino acid encoded by the nucleotide preceding the cut site in the direction of transcription. This is the nucleotide that corresponds to the 'Target Cut Length' in a CRISPick design file. We take 16 amino acids on either side of the cut site for a total sequence length of 33.
The get_aa_subseq_df from the targetfeat module will calculate these subsequences
from the complete amino acid sequences.
aa_subseq_df = get_aa_subseq_df(sg_designs=design_targ_df, aa_seq_df=aa_seq_df, width=16,
id_cols=id_cols)
aa_subseq_df.head()
.dataframe tbody tr th {
vertical-align: top;
}
.dataframe thead th {
text-align: right;
}
| Target Transcript | Target Total Length | Transcript Base | version | seq | molecule | desc | id | AA len | Target Cut Length | AA Index | Orientation | sgRNA Context Sequence | extended_seq | AA 0-Indexed | AA 0-Indexed padded | seq_start | seq_end | AA Subsequence | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | ENST00000259457.8 | 834 | ENST00000259457 | 3 | MAAVSVYAPPVGGFSFDNCRRNAVLEADFAKRGYKLPKVRKTGTTI... | protein | None | ENSP00000259457 | 277 | 191 | 64 | sense | TGGAGCAGATACAAGAGCAACTGAAGGGAT | -----------------MAAVSVYAPPVGGFSFDNCRRNAVLEADF... | 63 | 80 | 64 | 96 | GVVYKDGIVLGADTRATEGMVVADKNCSKIHFI |
| 1 | ENST00000259457.8 | 834 | ENST00000259457 | 3 | MAAVSVYAPPVGGFSFDNCRRNAVLEADFAKRGYKLPKVRKTGTTI... | protein | None | ENSP00000259457 | 277 | 137 | 46 | sense | CCGGAAAACTGGCACGACCATCGCTGGGGT | -----------------MAAVSVYAPPVGGFSFDNCRRNAVLEADF... | 45 | 62 | 46 | 78 | AKRGYKLPKVRKTGTTIAGVVYKDGIVLGADTR |
| 2 | ENST00000394249.8 | 1863 | ENST00000394249 | 3 | MRRSEVLAEESIVCLQKALNHLREIWELIGIPEDQRLQRTEVVKKH... | protein | None | ENSP00000377793 | 620 | 316 | 106 | sense | TAGAAAAAGATTTGCGCACCCAAGTGGAAT | -----------------MRRSEVLAEESIVCLQKALNHLREIWELI... | 105 | 122 | 106 | 138 | EEGETTILQLEKDLRTQVELMRKQKKERKQELK |
| 3 | ENST00000394249.8 | 1863 | ENST00000394249 | 3 | MRRSEVLAEESIVCLQKALNHLREIWELIGIPEDQRLQRTEVVKKH... | protein | None | ENSP00000377793 | 620 | 787 | 263 | antisense | TGGCCTTTGACCCAGACATAATGGTGGCCA | -----------------MRRSEVLAEESIVCLQKALNHLREIWELI... | 262 | 279 | 263 | 295 | WDRLQIPEEEREAVATIMSGSKAKVRKALQLEV |
| 4 | ENST00000361337.3 | 2298 | ENST00000361337 | 2 | MSGDHLHNDSQIEADFRLNDSHKHKDKHKDREHRHKEHKKEKDREK... | protein | None | ENSP00000354522 | 765 | 420 | 140 | antisense | AAATACTCACTCATCCTCATCTCGAGGTCT | -----------------MSGDHLHNDSQIEADFRLNDSHKHKDKHK... | 139 | 156 | 140 | 172 | GYFVPPKEDIKPLKRPRDEDDADYKPKKIKTED |
Lite Scores
You now have all the information you need to calculate "lite" Target Scores, which are less data intensive than complete
target scores, with the predict_target function from the predicttarg module.
lite_predictions = predict_target(design_df=design_df,
aa_subseq_df=aa_subseq_df)
design_df['Target Score Lite'] = lite_predictions
design_df.head()
/Users/peterdeweirdt/miniforge3/envs/test_rs3_v7/lib/python3.9/site-packages/sklearn/base.py:329: UserWarning: Trying to unpickle estimator SimpleImputer from version 1.0.dev0 when using version 1.0.2. This might lead to breaking code or invalid results. Use at your own risk. For more info please refer to:
https://scikit-learn.org/stable/modules/model_persistence.html#security-maintainability-limitations
warnings.warn(
/Users/peterdeweirdt/miniforge3/envs/test_rs3_v7/lib/python3.9/site-packages/sklearn/base.py:329: UserWarning: Trying to unpickle estimator Pipeline from version 1.0.dev0 when using version 1.0.2. This might lead to breaking code or invalid results. Use at your own risk. For more info please refer to:
https://scikit-learn.org/stable/modules/model_persistence.html#security-maintainability-limitations
warnings.warn(
/Users/peterdeweirdt/miniforge3/envs/test_rs3_v7/lib/python3.9/site-packages/sklearn/base.py:443: UserWarning: X has feature names, but SimpleImputer was fitted without feature names
warnings.warn(
.dataframe tbody tr th {
vertical-align: top;
}
.dataframe thead th {
text-align: right;
}
| Input | Quota | Target Taxon | Target Gene ID | Target Gene Symbol | Target Transcript | Target Reference Coords | Target Alias | CRISPR Mechanism | Target Domain | ... | Off-Target Rank Weight | Combined Rank | Preselected As | Matching Active Arrayed Oligos | Matching Arrayed Constructs | Pools Containing Matching Construct | Pick Order | Picking Round | Picking Notes | Target Score Lite | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | PSMB7 | 2 | 9606 | ENSG00000136930 | PSMB7 | ENST00000259457.8 | NaN | NaN | CRISPRko | CDS | ... | 1.0 | 7 | GCAGATACAAGAGCAACTGA | NaN | BRDN0004619103 | NaN | 1 | 0 | Preselected | -0.088666 |
| 1 | PSMB7 | 2 | 9606 | ENSG00000136930 | PSMB7 | ENST00000259457.8 | NaN | NaN | CRISPRko | CDS | ... | 1.0 | 48 | AAAACTGGCACGACCATCGC | NaN | NaN | NaN | 2 | 0 | Preselected | -0.013821 |
| 2 | PRC1 | 2 | 9606 | ENSG00000198901 | PRC1 | ENST00000394249.8 | NaN | NaN | CRISPRko | CDS | ... | 1.0 | 7 | AAAAGATTTGCGCACCCAAG | NaN | NaN | NaN | 1 | 0 | Preselected | 0.103036 |
| 3 | PRC1 | 2 | 9606 | ENSG00000198901 | PRC1 | ENST00000394249.8 | NaN | NaN | CRISPRko | CDS | ... | 1.0 | 8 | CTTTGACCCAGACATAATGG | NaN | NaN | NaN | 2 | 0 | Preselected | 0.160578 |
| 4 | TOP1 | 2 | 9606 | ENSG00000198900 | TOP1 | ENST00000361337.3 | NaN | NaN | CRISPRko | CDS | ... | 1.0 | 1 | NaN | NaN | BRDN0001486452 | NaN | 2 | 1 | NaN | 0.069597 |
5 rows × 61 columns
If you would like to calculate full target scores then follow the sections below.
Protein domain input
To calculate full target scores you will also need inputs for protein domains and conservation.
The protein domain input should have 16 binary columns for 16 different protein domain sources in addition to the
id_cols. The protein domain sources are 'Pfam', 'PANTHER', 'HAMAP', 'SuperFamily', 'TIGRfam', 'ncoils', 'Gene3D',
'Prosite_patterns', 'Seg', 'SignalP', 'TMHMM', 'MobiDBLite', 'PIRSF', 'PRINTS', 'Smart', 'Prosite_profiles'.
These columns should be kept in order when inputting for scoring.
In this example we will load the protein domain information from a parquet file, which was written
using write_transcript_data function in the targetdata module. You can also query transcript data on the fly,
by using the build_translation_overlap_df function. See the documentation for the predicttarg module for more
information on how to do this.
domain_df = pd.read_parquet('test_data/target_data/protein_domains.pq', engine='pyarrow',
filters=[[('Transcript Base', 'in', transcript_bases)]])
domain_df.head()
.dataframe tbody tr th {
vertical-align: top;
}
.dataframe thead th {
text-align: right;
}
| type | cigar_string | id | hit_end | feature_type | description | seq_region_name | end | hit_start | translation_id | interpro | hseqname | Transcript Base | align_type | start | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | Pfam | PF12465 | 36 | protein_feature | Proteasome beta subunit, C-terminal | ENSP00000259457 | 271 | 1 | 976188 | IPR024689 | PF12465 | ENST00000259457 | None | 235 | |
| 1 | Pfam | PF00227 | 190 | protein_feature | Proteasome, subunit alpha/beta | ENSP00000259457 | 221 | 2 | 976188 | IPR001353 | PF00227 | ENST00000259457 | None | 41 | |
| 2 | PRINTS | PR00141 | 0 | protein_feature | Peptidase T1A, proteasome beta-subunit | ENSP00000259457 | 66 | 0 | 976188 | IPR000243 | PR00141 | ENST00000259457 | None | 51 | |
| 3 | PRINTS | PR00141 | 0 | protein_feature | Peptidase T1A, proteasome beta-subunit | ENSP00000259457 | 182 | 0 | 976188 | IPR000243 | PR00141 | ENST00000259457 | None | 171 | |
| 4 | PRINTS | PR00141 | 0 | protein_feature | Peptidase T1A, proteasome beta-subunit | ENSP00000259457 | 193 | 0 | 976188 | IPR000243 | PR00141 | ENST00000259457 | None | 182 |
Now to transform the domain_df into a wide form for model input, we use the get_protein_domain_features function
from the targetfeat module.
domain_feature_df = get_protein_domain_features(design_targ_df, domain_df, id_cols=id_cols)
domain_feature_df.head()
.dataframe tbody tr th {
vertical-align: top;
}
.dataframe thead th {
text-align: right;
}
| sgRNA Context Sequence | Target Cut Length | Target Transcript | Orientation | Pfam | PANTHER | HAMAP | SuperFamily | TIGRfam | ncoils | Gene3D | Prosite_patterns | Seg | SignalP | TMHMM | MobiDBLite | PIRSF | PRINTS | Smart | Prosite_profiles | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | AAAAGAATGATGAAAAGACACCACAGGGAG | 244 | ENST00000610426.5 | sense | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 1 | AAAAGAGCCATGAATCTAAACATCAGGAAT | 640 | ENST00000223073.6 | sense | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| 2 | AAAAGCGCCAAATGGCCCGAGAATTGGGAG | 709 | ENST00000331923.9 | sense | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| 3 | AAACAGAAAAAGTTAAAATCACCAAGGTGT | 496 | ENST00000283882.4 | sense | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 4 | AAACAGATGGAAGATGCTTACCGGGGGACC | 132 | ENST00000393047.8 | sense | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
For input into the predict_target function, the domain_feature_df should have the id_cols as well as
columns for each of the 16 protein domain features.
Conservation input
Finally, for the full target model you need to calculate conservation features. The conservation features represent conservation across evolutionary time at the sgRNA cut site and are quantified using PhyloP scores. These scores are available for download by the UCSC genome browser for hg38 (phyloP100way), and mm39 (phyloP35way).
Within this package we query conservation scores using the UCSC genome browser's
REST API.
To get conservation scores, you can use the build_conservation_df function from the targetdata module.
Here we load conservation scores, which were written to parquet using the write_conservation_data function from the
targetdata module.
conservation_df = pd.read_parquet('test_data/target_data/conservation.pq', engine='pyarrow',
filters=[[('Transcript Base', 'in', transcript_bases)]])
conservation_df.head()
.dataframe tbody tr th {
vertical-align: top;
}
.dataframe thead th {
text-align: right;
}
| exon_id | genomic position | conservation | Transcript Base | target position | chromosome | genome | translation length | Target Transcript | Strand of Target | Target Total Length | ranked_conservation | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | ENSE00001866322 | 124415425.0 | 6.46189 | ENST00000259457 | 1 | 9 | hg38 | 277 | ENST00000259457.8 | - | 834 | 0.639089 |
| 1 | ENSE00001866322 | 124415424.0 | 7.48071 | ENST00000259457 | 2 | 9 | hg38 | 277 | ENST00000259457.8 | - | 834 | 0.686451 |
| 2 | ENSE00001866322 | 124415423.0 | 6.36001 | ENST00000259457 | 3 | 9 | hg38 | 277 | ENST00000259457.8 | - | 834 | 0.622902 |
| 3 | ENSE00001866322 | 124415422.0 | 6.36001 | ENST00000259457 | 4 | 9 | hg38 | 277 | ENST00000259457.8 | - | 834 | 0.622902 |
| 4 | ENSE00001866322 | 124415421.0 | 8.09200 | ENST00000259457 | 5 | 9 | hg38 | 277 | ENST00000259457.8 | - | 834 | 0.870504 |
We normalize conservation scores to a within-gene percent rank, in the 'ranked_conservation' column, in order to make scores comparable across genes and genomes. Note that a rank of 0 indicates the least conserved nucleotide and a rank of 1 indicates the most conserved.
To featurize the conservation scores, we average across a window of 4 and 32 nucleotides centered around the nucleotide preceding the cut site in the direction of transcription. Note that this nucleotide is the 2nd nucleotide in the window of 4 and the 16th nucleotide in the window of 32.
We use the get_conservation_features function from the targetfeat module to get these features from the
conservation_df.
For the predict_targ function, we need the id_cols and the columns 'cons_4' and 'cons_32' in the
conservation_feature_df.
conservation_feature_df = get_conservation_features(design_targ_df, conservation_df,
small_width=2, large_width=16,
conservation_column='ranked_conservation',
id_cols=id_cols)
conservation_feature_df
.dataframe tbody tr th {
vertical-align: top;
}
.dataframe thead th {
text-align: right;
}
| sgRNA Context Sequence | Target Cut Length | Target Transcript | Orientation | cons_4 | cons_32 | |
|---|---|---|---|---|---|---|
| 0 | AAAAGAATGATGAAAAGACACCACAGGGAG | 244 | ENST00000610426.5 | sense | 0.218231 | 0.408844 |
| 1 | AAAAGAGCCATGAATCTAAACATCAGGAAT | 640 | ENST00000223073.6 | sense | 0.129825 | 0.278180 |
| 2 | AAAAGCGCCAAATGGCCCGAGAATTGGGAG | 709 | ENST00000331923.9 | sense | 0.470906 | 0.532305 |
| 3 | AAACAGAAAAAGTTAAAATCACCAAGGTGT | 496 | ENST00000283882.4 | sense | 0.580556 | 0.602708 |
| 4 | AAACAGATGGAAGATGCTTACCGGGGGACC | 132 | ENST00000393047.8 | sense | 0.283447 | 0.414293 |
| ... | ... | ... | ... | ... | ... | ... |
| 395 | TTTGATTGCATTAAGGTTGGACTCTGGATT | 246 | ENST00000249269.9 | sense | 0.580612 | 0.618707 |
| 396 | TTTGCCCACAGCTCCAAAGCATCGCGGAGA | 130 | ENST00000227618.8 | sense | 0.323770 | 0.416368 |
| 397 | TTTTACAGTGCGATGTATGATGTATGGCTT | 119 | ENST00000338366.6 | sense | 0.788000 | 0.537417 |
| 398 | TTTTGGATCTCGTAGTGATTCAAGAGGGAA | 233 | ENST00000629496.3 | sense | 0.239630 | 0.347615 |
| 399 | TTTTTGTTACTACAGGTTCGCTGCTGGGAA | 201 | ENST00000395840.6 | sense | 0.693767 | 0.639044 |
400 rows × 6 columns
Full Target Scores
In order to calculate Target Scores you must input the feature matrices and design_df to the predict_target
function from the predicttarg module.
target_predictions = predict_target(design_df=design_df,
aa_subseq_df=aa_subseq_df,
domain_feature_df=domain_feature_df,
conservation_feature_df=conservation_feature_df,
id_cols=id_cols)
design_df['Target Score'] = target_predictions
design_df.head()
/Users/peterdeweirdt/miniforge3/envs/test_rs3_v7/lib/python3.9/site-packages/sklearn/base.py:329: UserWarning: Trying to unpickle estimator SimpleImputer from version 1.0.dev0 when using version 1.0.2. This might lead to breaking code or invalid results. Use at your own risk. For more info please refer to:
https://scikit-learn.org/stable/modules/model_persistence.html#security-maintainability-limitations
warnings.warn(
/Users/peterdeweirdt/miniforge3/envs/test_rs3_v7/lib/python3.9/site-packages/sklearn/base.py:329: UserWarning: Trying to unpickle estimator Pipeline from version 1.0.dev0 when using version 1.0.2. This might lead to breaking code or invalid results. Use at your own risk. For more info please refer to:
https://scikit-learn.org/stable/modules/model_persistence.html#security-maintainability-limitations
warnings.warn(
/Users/peterdeweirdt/miniforge3/envs/test_rs3_v7/lib/python3.9/site-packages/sklearn/base.py:443: UserWarning: X has feature names, but SimpleImputer was fitted without feature names
warnings.warn(
.dataframe tbody tr th {
vertical-align: top;
}
.dataframe thead th {
text-align: right;
}
| Input | Quota | Target Taxon | Target Gene ID | Target Gene Symbol | Target Transcript | Target Reference Coords | Target Alias | CRISPR Mechanism | Target Domain | ... | Combined Rank | Preselected As | Matching Active Arrayed Oligos | Matching Arrayed Constructs | Pools Containing Matching Construct | Pick Order | Picking Round | Picking Notes | Target Score Lite | Target Score | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | PSMB7 | 2 | 9606 | ENSG00000136930 | PSMB7 | ENST00000259457.8 | NaN | NaN | CRISPRko | CDS | ... | 7 | GCAGATACAAGAGCAACTGA | NaN | BRDN0004619103 | NaN | 1 | 0 | Preselected | -0.088666 | 0.110229 |
| 1 | PSMB7 | 2 | 9606 | ENSG00000136930 | PSMB7 | ENST00000259457.8 | NaN | NaN | CRISPRko | CDS | ... | 48 | AAAACTGGCACGACCATCGC | NaN | NaN | NaN | 2 | 0 | Preselected | -0.013821 | 0.018766 |
| 2 | PRC1 | 2 | 9606 | ENSG00000198901 | PRC1 | ENST00000394249.8 | NaN | NaN | CRISPRko | CDS | ... | 7 | AAAAGATTTGCGCACCCAAG | NaN | NaN | NaN | 1 | 0 | Preselected | 0.103036 | 0.127005 |
| 3 | PRC1 | 2 | 9606 | ENSG00000198901 | PRC1 | ENST00000394249.8 | NaN | NaN | CRISPRko | CDS | ... | 8 | CTTTGACCCAGACATAATGG | NaN | NaN | NaN | 2 | 0 | Preselected | 0.160578 | -0.035591 |
| 4 | TOP1 | 2 | 9606 | ENSG00000198900 | TOP1 | ENST00000361337.3 | NaN | NaN | CRISPRko | CDS | ... | 1 | NaN | NaN | BRDN0001486452 | NaN | 2 | 1 | NaN | 0.069597 | -0.154246 |
5 rows × 62 columns
Target Scores can be added directly to the sequence scores for your final Rule Set 3 predictions.
Predict Function
If you don't want to generate the target matrices themselves, you can use the predict function from
the predict module.
from rs3.predict import predict
import matplotlib.pyplot as plt
import gpplot
import seaborn as sns
Preloaded data
In this first example with the predict function, we calculate predictions for GeckoV2 sgRNAs.
In this example the amino acid sequences, protein domains and conservation scores were prequeried using the
write_transcript_data and write_conservation_data functions from the targetdata module.
Pre-querying these data can be helpful for large scale design runs.
You can also use the predict function without pre-querying and calculate
scores on the fly. You can see an example of this in the next section.
The predict function allows for parallel computation
for querying databases (n_jobs_min) and featurizing sgRNAs (n_jobs_max).
We recommend keeping n_jobs_min set to 1 or 2, as the APIs limit the amount of queries per hour.
design_df = pd.read_table('test_data/sgrna-designs.txt')
import multiprocessing
max_n_jobs = multiprocessing.cpu_count()
scored_designs = predict(design_df, tracr=['Hsu2013', 'Chen2013'], target=True,
n_jobs_min=2, n_jobs_max=max_n_jobs,
aa_seq_file='./test_data/target_data/aa_seqs.pq',
domain_file='./test_data/target_data/protein_domains.pq',
conservatin_file='./test_data/target_data/conservation.pq',
lite=False)
scored_designs.head()
Calculating sequence-based features
100%|██████████| 400/400 [00:01<00:00, 226.77it/s]
Calculating sequence-based features
100%|██████████| 400/400 [00:00<00:00, 2734.69it/s]
/Users/peterdeweirdt/miniforge3/envs/test_rs3_v7/lib/python3.9/site-packages/sklearn/base.py:329: UserWarning: Trying to unpickle estimator SimpleImputer from version 1.0.dev0 when using version 1.0.2. This might lead to breaking code or invalid results. Use at your own risk. For more info please refer to:
https://scikit-learn.org/stable/modules/model_persistence.html#security-maintainability-limitations
warnings.warn(
/Users/peterdeweirdt/miniforge3/envs/test_rs3_v7/lib/python3.9/site-packages/sklearn/base.py:329: UserWarning: Trying to unpickle estimator Pipeline from version 1.0.dev0 when using version 1.0.2. This might lead to breaking code or invalid results. Use at your own risk. For more info please refer to:
https://scikit-learn.org/stable/modules/model_persistence.html#security-maintainability-limitations
warnings.warn(
/Users/peterdeweirdt/miniforge3/envs/test_rs3_v7/lib/python3.9/site-packages/sklearn/base.py:443: UserWarning: X has feature names, but SimpleImputer was fitted without feature names
warnings.warn(
.dataframe tbody tr th {
vertical-align: top;
}
.dataframe thead th {
text-align: right;
}
| Input | Quota | Target Taxon | Target Gene ID | Target Gene Symbol | Target Transcript | Target Reference Coords | Target Alias | CRISPR Mechanism | Target Domain | ... | Picking Round | Picking Notes | RS3 Sequence Score (Hsu2013 tracr) | RS3 Sequence Score (Chen2013 tracr) | AA Index | Transcript Base | Missing conservation information | Target Score | RS3 Sequence (Hsu2013 tracr) + Target Score | RS3 Sequence (Chen2013 tracr) + Target Score | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | PSMB7 | 2 | 9606 | ENSG00000136930 | PSMB7 | ENST00000259457.8 | NaN | NaN | CRISPRko | CDS | ... | 0 | Preselected | 0.787640 | 0.559345 | 64 | ENST00000259457 | False | 0.110229 | 0.897869 | 0.669574 |
| 1 | PSMB7 | 2 | 9606 | ENSG00000136930 | PSMB7 | ENST00000259457.8 | NaN | NaN | CRISPRko | CDS | ... | 0 | Preselected | -0.294126 | -0.181437 | 46 | ENST00000259457 | False | 0.018766 | -0.275359 | -0.162671 |
| 2 | PRC1 | 2 | 9606 | ENSG00000198901 | PRC1 | ENST00000394249.8 | NaN | NaN | CRISPRko | CDS | ... | 0 | Preselected | -0.043418 | -0.220434 | 106 | ENST00000394249 | False | 0.127005 | 0.083588 | -0.093429 |
| 3 | PRC1 | 2 | 9606 | ENSG00000198901 | PRC1 | ENST00000394249.8 | NaN | NaN | CRISPRko | CDS | ... | 0 | Preselected | 0.759256 | 0.453469 | 263 | ENST00000394249 | False | -0.035591 | 0.723665 | 0.417878 |
| 4 | TOP1 | 2 | 9606 | ENSG00000198900 | TOP1 | ENST00000361337.3 | NaN | NaN | CRISPRko | CDS | ... | 1 | NaN | 0.424001 | -0.197035 | 140 | ENST00000361337 | False | -0.154246 | 0.269755 | -0.351282 |
5 rows × 68 columns
Here are the details for the keyword arguments of the above function
tracr- tracr to calculate scores for. If a list is supplied instead of a string, scores will be calculated for both tracrstarget- boolean indicating whether to calculate target scoresn_jobs_min,n_jobs_max- number of cpus to use for parallel computationaa_seq_file,domain_file,conservatin_file- precalculated parquet files. Optional inputs as these features can also be calculated on the flylite- boolean indicating whether to calculate lite target scores
By listing both tracrRNAs tracr=['Hsu2013', 'Chen2013'] and setting target=True,
we calculate 5 unique scores: one sequence score for each tracr, the target score,
and the sequence scores plus the target score.
We can compare these predictions against the observed activity from GeckoV2
gecko_activity = pd.read_csv('test_data/Aguirre2016_activity.csv')
gecko_activity.head()
.dataframe tbody tr th {
vertical-align: top;
}
.dataframe thead th {
text-align: right;
}
| sgRNA Sequence | sgRNA Context Sequence | Target Gene Symbol | Target Cut % | avg_mean_centered_neg_lfc | |
|---|---|---|---|---|---|
| 0 | AAAAAACTTACCCCTTTGAC | AAAAAAAAAACTTACCCCTTTGACTGGCCA | CPSF6 | 22.2 | -1.139819 |
| 1 | AAAAACATTATCATTGAGCC | TGGCAAAAACATTATCATTGAGCCTGGATT | SKA3 | 62.3 | -0.793055 |
| 2 | AAAAAGAGATTGTCAAATCA | TATGAAAAAGAGATTGTCAAATCAAGGTAG | AQR | 3.8 | 0.946453 |
| 3 | AAAAAGCATCTCTAGAAATA | TTCAAAAAAGCATCTCTAGAAATATGGTCC | ZNHIT6 | 61.7 | -0.429590 |
| 4 | AAAAAGCGAGATACCCGAAA | AAAAAAAAAGCGAGATACCCGAAAAGGCAG | ABCF1 | 9.4 | 0.734196 |
gecko_activity_scores = (gecko_activity.merge(scored_designs,
how='inner',
on=['sgRNA Sequence', 'sgRNA Context Sequence',
'Target Gene Symbol', 'Target Cut %']))
gecko_activity_scores.head()
.dataframe tbody tr th {
vertical-align: top;
}
.dataframe thead th {
text-align: right;
}
| sgRNA Sequence | sgRNA Context Sequence | Target Gene Symbol | Target Cut % | avg_mean_centered_neg_lfc | Input | Quota | Target Taxon | Target Gene ID | Target Transcript | ... | Picking Round | Picking Notes | RS3 Sequence Score (Hsu2013 tracr) | RS3 Sequence Score (Chen2013 tracr) | AA Index | Transcript Base | Missing conservation information | Target Score | RS3 Sequence (Hsu2013 tracr) + Target Score | RS3 Sequence (Chen2013 tracr) + Target Score | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | AAAACTGGCACGACCATCGC | CCGGAAAACTGGCACGACCATCGCTGGGGT | PSMB7 | 16.4 | -1.052943 | PSMB7 | 2 | 9606 | ENSG00000136930 | ENST00000259457.8 | ... | 0 | Preselected | -0.294126 | -0.181437 | 46 | ENST00000259457 | False | 0.018766 | -0.275359 | -0.162671 |
| 1 | AAAAGATTTGCGCACCCAAG | TAGAAAAAGATTTGCGCACCCAAGTGGAAT | PRC1 | 17.0 | 0.028674 | PRC1 | 2 | 9606 | ENSG00000198901 | ENST00000394249.8 | ... | 0 | Preselected | -0.043418 | -0.220434 | 106 | ENST00000394249 | False | 0.127005 | 0.083588 | -0.093429 |
| 2 | AAAAGTCCAAGCATAGCAAC | CGGGAAAAGTCCAAGCATAGCAACAGGTAA | TOP1 | 6.5 | 0.195309 | TOP1 | 2 | 9606 | ENSG00000198900 | ENST00000361337.3 | ... | 0 | Preselected | -0.294127 | -0.022951 | 50 | ENST00000361337 | False | -0.137369 | -0.431496 | -0.160320 |
| 3 | AAAGAAGCCTCAACTTCGTC | AGCGAAAGAAGCCTCAACTTCGTCTGGAGA | CENPW | 37.5 | -1.338209 | CENPW | 2 | 9606 | ENSG00000203760 | ENST00000368328.5 | ... | 0 | Preselected | -0.667399 | -0.308794 | 34 | ENST00000368328 | False | 0.120044 | -0.547355 | -0.188750 |
| 4 | AAAGTGTGCTTTGTTGGAGA | TACTAAAGTGTGCTTTGTTGGAGATGGCTT | NSA2 | 60.0 | -0.175219 | NSA2 | 2 | 9606 | ENSG00000164346 | ENST00000610426.5 | ... | 0 | Preselected | -0.402220 | -0.622492 | 157 | ENST00000610426 | False | 0.047223 | -0.354996 | -0.575268 |
5 rows × 69 columns
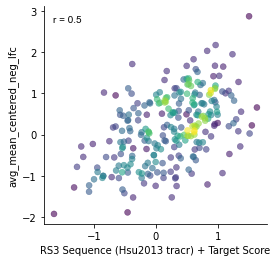
Since GeckoV2 was screened with the tracrRNA from Hsu et al. 2013, we'll use these scores sequence scores a part of our final prediction.
plt.subplots(figsize=(4,4))
gpplot.point_densityplot(gecko_activity_scores, y='avg_mean_centered_neg_lfc',
x='RS3 Sequence (Hsu2013 tracr) + Target Score')
gpplot.add_correlation(gecko_activity_scores, y='avg_mean_centered_neg_lfc',
x='RS3 Sequence (Hsu2013 tracr) + Target Score')
sns.despine()
Predictions on the fly
You can also make predictions without pre-querying the target data. Here we use example designs for BCL2L1, MCL1 and EEF2.
design_df = pd.read_table('test_data/sgrna-designs_BCL2L1_MCL1_EEF2.txt')
scored_designs = predict(design_df,
tracr=['Hsu2013', 'Chen2013'], target=True,
n_jobs_min=2, n_jobs_max=8,
lite=False)
scored_designs
Calculating sequence-based features
100%|██████████| 849/849 [00:00<00:00, 2397.41it/s]
Calculating sequence-based features
100%|██████████| 849/849 [00:00<00:00, 3400.56it/s]
Getting amino acid sequences
100%|██████████| 1/1 [00:00<00:00, 4.01it/s]
Getting protein domains
100%|██████████| 3/3 [00:00<00:00, 2714.76it/s]
Getting conservation
100%|██████████| 3/3 [00:00<00:00, 29.72it/s]
/Users/peterdeweirdt/miniforge3/envs/test_rs3_v7/lib/python3.9/site-packages/sklearn/base.py:329: UserWarning: Trying to unpickle estimator SimpleImputer from version 1.0.dev0 when using version 1.0.2. This might lead to breaking code or invalid results. Use at your own risk. For more info please refer to:
https://scikit-learn.org/stable/modules/model_persistence.html#security-maintainability-limitations
warnings.warn(
/Users/peterdeweirdt/miniforge3/envs/test_rs3_v7/lib/python3.9/site-packages/sklearn/base.py:329: UserWarning: Trying to unpickle estimator Pipeline from version 1.0.dev0 when using version 1.0.2. This might lead to breaking code or invalid results. Use at your own risk. For more info please refer to:
https://scikit-learn.org/stable/modules/model_persistence.html#security-maintainability-limitations
warnings.warn(
/Users/peterdeweirdt/miniforge3/envs/test_rs3_v7/lib/python3.9/site-packages/sklearn/base.py:443: UserWarning: X has feature names, but SimpleImputer was fitted without feature names
warnings.warn(
.dataframe tbody tr th {
vertical-align: top;
}
.dataframe thead th {
text-align: right;
}
| Input | Quota | Target Taxon | Target Gene ID | Target Gene Symbol | Target Transcript | Target Alias | CRISPR Mechanism | Target Domain | Reference Sequence | ... | Picking Round | Picking Notes | RS3 Sequence Score (Hsu2013 tracr) | RS3 Sequence Score (Chen2013 tracr) | AA Index | Transcript Base | Missing conservation information | Target Score | RS3 Sequence (Hsu2013 tracr) + Target Score | RS3 Sequence (Chen2013 tracr) + Target Score | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | EEF2 | 5 | 9606 | ENSG00000167658 | EEF2 | ENST00000309311.7 | NaN | CRISPRko | CDS | NC_000019.10 | ... | NaN | Outside Target Window: 5-65% | 0.907809 | 0.769956 | 666 | ENST00000309311 | False | -0.143972 | 0.763837 | 0.625984 |
| 1 | EEF2 | 5 | 9606 | ENSG00000167658 | EEF2 | ENST00000309311.7 | NaN | CRISPRko | CDS | NC_000019.10 | ... | NaN | BsmBI:CGTCTC; Outside Target Window: 5-65% | 0.171870 | 0.040419 | 581 | ENST00000309311 | False | -0.027326 | 0.144543 | 0.013093 |
| 2 | EEF2 | 5 | 9606 | ENSG00000167658 | EEF2 | ENST00000309311.7 | NaN | CRISPRko | CDS | NC_000019.10 | ... | 1.0 | NaN | 1.393513 | 0.577732 | 107 | ENST00000309311 | False | -0.002082 | 1.391430 | 0.575650 |
| 3 | EEF2 | 5 | 9606 | ENSG00000167658 | EEF2 | ENST00000309311.7 | NaN | CRISPRko | CDS | NC_000019.10 | ... | 1.0 | NaN | 0.904446 | 0.008390 | 406 | ENST00000309311 | False | 0.080430 | 0.984876 | 0.088820 |
| 4 | EEF2 | 5 | 9606 | ENSG00000167658 | EEF2 | ENST00000309311.7 | NaN | CRISPRko | CDS | NC_000019.10 | ... | 1.0 | NaN | 0.831087 | 0.361594 | 546 | ENST00000309311 | False | 0.025749 | 0.856836 | 0.387343 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 844 | MCL1 | 5 | 9606 | ENSG00000143384 | MCL1 | ENST00000369026.3 | NaN | CRISPRko | CDS | NC_000001.11 | ... | NaN | Off-target Match Bin I matches > 3; Spacing Vi... | -0.792918 | -0.663881 | 52 | ENST00000369026 | False | -0.136403 | -0.929321 | -0.800284 |
| 845 | MCL1 | 5 | 9606 | ENSG00000143384 | MCL1 | ENST00000369026.3 | NaN | CRISPRko | CDS | NC_000001.11 | ... | NaN | Outside Target Window: 5-65%; poly(T):TTTT | -1.920374 | -1.819985 | 5 | ENST00000369026 | False | -0.003507 | -1.923881 | -1.823491 |
| 846 | MCL1 | 5 | 9606 | ENSG00000143384 | MCL1 | ENST00000369026.3 | NaN | CRISPRko | CDS | NC_000001.11 | ... | NaN | Spacing Violation: Too close to earlier pick a... | -1.101303 | -1.295640 | 24 | ENST00000369026 | False | -0.148665 | -1.249969 | -1.444305 |
| 847 | MCL1 | 5 | 9606 | ENSG00000143384 | MCL1 | ENST00000369026.3 | NaN | CRISPRko | CDS | NC_000001.11 | ... | NaN | Spacing Violation: Too close to earlier pick a... | -0.617431 | -0.621436 | 30 | ENST00000369026 | False | -0.145388 | -0.762819 | -0.766825 |
| 848 | MCL1 | 5 | 9606 | ENSG00000143384 | MCL1 | ENST00000369026.3 | NaN | CRISPRko | CDS | NC_000001.11 | ... | NaN | On-Target Efficacy Score < 0.2; Spacing Violat... | -0.586811 | -0.664130 | 30 | ENST00000369026 | False | -0.145388 | -0.732200 | -0.809518 |
849 rows × 61 columns
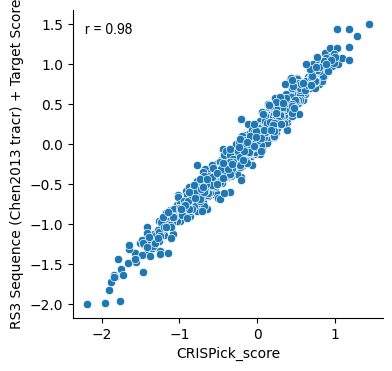
crispick_designs = pd.read_table('test_data/CRISPick_reference_designs.txt')
merged_designs = (scored_designs
.merge(crispick_designs[['sgRNA Cut Position (1-based)', 'sgRNA Sequence', 'On-Target Efficacy Score']]
.rename(columns={'On-Target Efficacy Score': 'CRISPick_score'}), how='inner',
on=['sgRNA Cut Position (1-based)', 'sgRNA Sequence']))
plt.subplots(figsize=(4,4))
gpplot.add_correlation(data=merged_designs, x='CRISPick_score', y='RS3 Sequence (Chen2013 tracr) + Target Score')
sns.scatterplot(data=merged_designs, x='CRISPick_score', y='RS3 Sequence (Chen2013 tracr) + Target Score')
sns.despine()
We see that the predict function is querying the target data in addition to making predictions.
4 maintainers
Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file rs3-0.0.18.tar.gz.
File metadata
- Download URL: rs3-0.0.18.tar.gz
- Upload date:
- Size: 6.1 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.10.18
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
6edd942b3e266b3e838ddecc5b7b387c911c58c6024da06d5908ceb999827308
|
|
| MD5 |
0dbefb9afc61306be7048cefa3478312
|
|
| BLAKE2b-256 |
a56d224b0a7039b8ffda89a355590cf8e39b4f232228484399b14d974f50f474
|
File details
Details for the file rs3-0.0.18-py3-none-any.whl.
File metadata
- Download URL: rs3-0.0.18-py3-none-any.whl
- Upload date:
- Size: 6.2 MB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.10.18
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
1c10ac0d6be46c2e1a71a3bd400d2b6fefcc7dff19de06c83e0854f16dfd5d2f
|
|
| MD5 |
0d5958b10b904781aa3fcd097a5d99a0
|
|
| BLAKE2b-256 |
3ba30fc328b2852fabcdf0ef06a3d96af2ecb35c20946d8be0b941b7d3854e38
|









