RNA velocity generalized through dynamical modeling
Verified details
These details have been verified by PyPIProject links
Owner
GitHub Statistics
Maintainers


Project description



scVelo - RNA velocity generalized through dynamical modeling
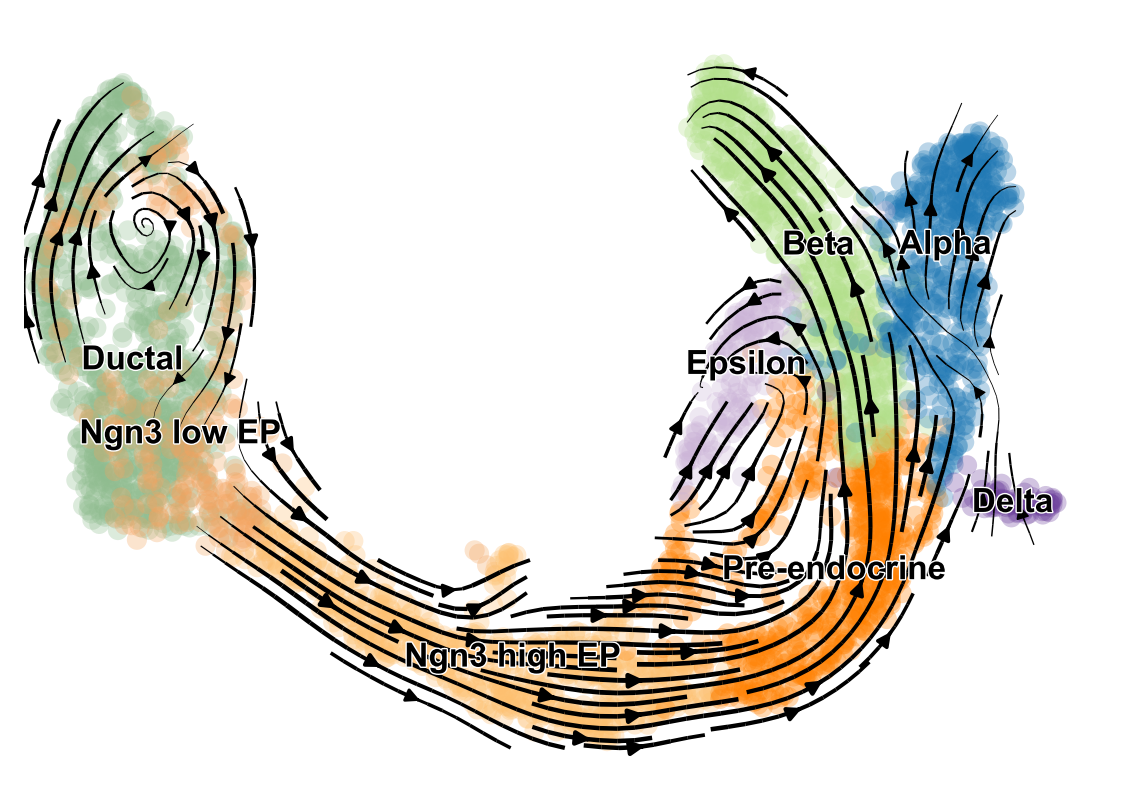
scVelo is a scalable toolkit for RNA velocity analysis in single cells; RNA velocity enables the recovery of directed dynamic information by leveraging splicing kinetics 1. scVelo collects different methods for inferring RNA velocity using an expectation-maximization framework 2, deep generative modeling 3, or metabolically labeled transcripts4.
scVelo's key applications
- estimate RNA velocity to study cellular dynamics.
- identify putative driver genes and regimes of regulatory changes.
- infer a latent time to reconstruct the temporal sequence of transcriptomic events.
- estimate reaction rates of transcription, splicing and degradation.
- use statistical tests, e.g., to detect different kinetics regimes.
Citing scVelo
If you include or rely on scVelo when publishing research, please adhere to the following citation guide:
EM and steady-state model
If you use the EM (dynamical) or steady-state model, cite
@article{Bergen2020,
title = {Generalizing RNA velocity to transient cell states through dynamical modeling},
volume = {38},
ISSN = {1546-1696},
url = {http://dx.doi.org/10.1038/s41587-020-0591-3},
DOI = {10.1038/s41587-020-0591-3},
number = {12},
journal = {Nature Biotechnology},
publisher = {Springer Science and Business Media LLC},
author = {Bergen, Volker and Lange, Marius and Peidli, Stefan and Wolf, F. Alexander and Theis, Fabian J.},
year = {2020},
month = aug,
pages = {1408–1414}
}
RNA velocity inference through metabolic labeling information
If you use the implemented method for estimating RNA velocity from metabolic labeling information, cite
@article{Weiler2024,
author = {Weiler, Philipp and Lange, Marius and Klein, Michal and Pe'er, Dana and Theis, Fabian},
publisher = {Springer Science and Business Media LLC},
url = {http://dx.doi.org/10.1038/s41592-024-02303-9},
doi = {10.1038/s41592-024-02303-9},
issn = {1548-7105},
journal = {Nature Methods},
month = jun,
number = {7},
pages = {1196--1205},
title = {CellRank 2: unified fate mapping in multiview single-cell data},
volume = {21},
year = {2024},
}
Support
Found a bug or would like to see a feature implemented? Feel free to submit an issue. Have a question or would like to start a new discussion? Head over to GitHub discussions. Your help to improve scVelo is highly appreciated. For further information visit scvelo.org.
Project details
Verified details
These details have been verified by PyPIProject links
Owner
GitHub Statistics
Maintainers
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file scvelo-0.3.4.tar.gz.
File metadata
- Download URL: scvelo-0.3.4.tar.gz
- Upload date:
- Size: 27.3 MB
- Tags: Source
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
9480c111105a33638152ea34d61e9e92aa1abc251bc31158601a8678ee4886b9
|
|
| MD5 |
011b45fb2eefe3e448e5a1740959fb8c
|
|
| BLAKE2b-256 |
4ed552d0a783a321e506bdf8e65691985c4af509a5b848c923746f94cfe25207
|
Provenance
The following attestation bundles were made for scvelo-0.3.4.tar.gz:
Publisher:
release.yml on theislab/scvelo
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
scvelo-0.3.4.tar.gz -
Subject digest:
9480c111105a33638152ea34d61e9e92aa1abc251bc31158601a8678ee4886b9 - Sigstore transparency entry: 988216631
- Sigstore integration time:
-
Permalink:
theislab/scvelo@9b6e94647654eca1c4a38b08a21f59df8a4ae6a6 -
Branch / Tag:
refs/tags/v0.3.4 - Owner: https://github.com/theislab
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
release.yml@9b6e94647654eca1c4a38b08a21f59df8a4ae6a6 -
Trigger Event:
push
-
Statement type:
File details
Details for the file scvelo-0.3.4-py3-none-any.whl.
File metadata
- Download URL: scvelo-0.3.4-py3-none-any.whl
- Upload date:
- Size: 191.2 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? Yes
- Uploaded via: twine/6.1.0 CPython/3.13.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
e54102b7a34241b8952728af49350c2710bd3873e37686a746f1d1e3e6314b1a
|
|
| MD5 |
c909c6a492b280bd81a0e983ac716992
|
|
| BLAKE2b-256 |
9536f3f611bcbe4af61ed30c8de86c2ba7e3727567b7c968fd9bebada82e9125
|
Provenance
The following attestation bundles were made for scvelo-0.3.4-py3-none-any.whl:
Publisher:
release.yml on theislab/scvelo
-
Statement:
-
Statement type:
https://in-toto.io/Statement/v1 -
Predicate type:
https://docs.pypi.org/attestations/publish/v1 -
Subject name:
scvelo-0.3.4-py3-none-any.whl -
Subject digest:
e54102b7a34241b8952728af49350c2710bd3873e37686a746f1d1e3e6314b1a - Sigstore transparency entry: 988216775
- Sigstore integration time:
-
Permalink:
theislab/scvelo@9b6e94647654eca1c4a38b08a21f59df8a4ae6a6 -
Branch / Tag:
refs/tags/v0.3.4 - Owner: https://github.com/theislab
-
Access:
public
-
Token Issuer:
https://token.actions.githubusercontent.com -
Runner Environment:
github-hosted -
Publication workflow:
release.yml@9b6e94647654eca1c4a38b08a21f59df8a4ae6a6 -
Trigger Event:
push
-
Statement type:







