Python port of the R Bioconductor `seqlogo` package

Project description



seqlogo
Python port of Bioconductor's seqLogo served by WebLogo
Overview
In the field of bioinformatics, a common task is to look for sequence motifs at different sites along the genome or within a protein sequence. One aspect of this analysis involves creating a variant of a Position Matrix (PM): Position Frequency Matrix (PFM), Position Probability Matrix (PPM), and Position Weight Matrix (PWM). The formal format for a PWM file can be found here.
Specification
A PM file can be just a plain text, whitespace delimited matrix, such that the number of columns
matches the number of letters in your desired alphabet and the number of rows is the number of positions
in your sequence. Any comment lines that start with # will be skipped.
Note: TRANSFAC matrix and MEME Motif formats are not directly supported.

Where 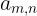
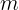
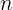
This is often generated in a frequentist fashion. If a pipeline tallies all observed letters at each position, this is called a Position Frequency Matrix (PFM).
The PFM can be converted to a PPM in a straight-forward manner, creating a matrix that for any given position and letter, the probability of that letter at that position is reported.
A PWM is the PPM converted into log-likelihood. Pseudocounts can be applied to prevent probabilities of 0 from turing into -inf in the conversion process. Lastly, each position's log-likelihood is corrected for some background probability for every given letter in the selected alphabet.
Features
-
seqlogocan use any PM as entry points for analysis (from a file or in array formats) and, subsequently, plot the sequence logos. -
seqlogowas written to support BIOINF 529 :Bioinformatics Concepts and Algorithms at the University of Michigan in the Department of Computational Medicine & Bioinformatics. -
seqlogoattempts to blend the user-friendly api of Bioconductor's seqLogo and the rendering power of the WebLogoPython API. -
seqlogosupports the following alphabets:Alphabet name Alphabet Letters "DNA""ACGT""reduced DNA""ACGTN-""ambig DNA""ACGTRYSWKMBDHVN-""RNA""ACGU""reduced RNA""ACGUN-""ambig RNA""ACGURYSWKMBDHVN-""AA""ACDEFGHIKLMNPQRSTVWY""reduced AA""ACDEFGHIKLMNPQRSTVWYX*-""ambig AA""ACDEFGHIKLMNOPQRSTUVWYBJZX*-"(Bolded alphabet names are the most commonly used) -
seqlogocan also render sequence logos in a number of formats:"svg""eps""pdf""jpeg""png"(default)
-
All plots can be rendered in 4 different sizes:
"small": 3.54" wide"medium": 5" wide"large": 7.25" wide"xlarge": 10.25" wide
Note: all sizes taken from this publication guide from Science Magazine.
Recommended settings:
- For best results, implement
seqlogowithin a IPython/Jupyter environment (for inline plotting purposes). - Initially written for Python 3.7, but has shown to work in versions 3.5+ (Python 2.7 is not supported)
Setup
Minimal Requirements:
numpypandasweblogo
Note: it is strongly encouraged that jupyter is installed as well.
conda environment:
To produce the ideal virtual environment that will run seqlogo on a conda-based
build, clone the repo or download the environment.yml within the repo. Then run the following
command:
$ conda env create -f environment.yml
Installation
To install using pip: (recommended)
$ pip install seqlogo
To install using conda
$ conda install -c bioconda seqlogo
Or install from GitHub directly
$ pip install git+https://github.com/betteridiot/seqlogo.git#egg=seqlogo
Quickstart
Importing
import numpy as np
import pandas as pd
import seqlogo
Generate some PM data (without frequency data)
For many demonstrations that speak to PWMs, they are often started with PPM data.
Many packages preclude sequence logo generation from this entry point. However,
seqlogo can handle it just fine. One point to make though is that if no count
data is provided, seqlogo just generates the PFM data by multiplying the
probabilities by 100. This is only for weblogolib compatability.
# Setting seed for demonstration purposes
>>> np.random.seed(42)
# Making a fake PPM
>>> random_ppm = np.random.dirichlet(np.ones(4), size=6)
>>> ppm = seqlogo.Ppm(random_ppm)
>>> ppm
A C G T
0 0.082197 0.527252 0.230641 0.159911
1 0.070375 0.070363 0.024826 0.834435
2 0.161962 0.216972 0.003665 0.617401
3 0.735638 0.098290 0.082638 0.083434
4 0.179898 0.368931 0.280463 0.170708
5 0.498510 0.079138 0.182004 0.240349
Generate some frequency data and convert to PWM
Sometimes the user has frequency data instead of PWM. To construct a Pwm instance
that automatically computes Information Content and PWM values, the user can use
the seqlogo.pfm2pwm() function.
# Setting seed for demonstration purposes
>>> np.random.seed(42)
# Making some fake Position Frequency Data (PFM)
>>> pfm = pd.DataFrame(np.random.randint(0, 36, size=(8, 4)))
# Convert to Position Weight Matrix (PWM)
>>> pwm = seqlogo.pfm2pwm(pfm)
>>> pwm
A C G T
0 0.698830 -0.301170 -1.301170 0.213404
1 0.263034 0.552541 -0.584962 -0.584962
2 0.148523 0.754244 0.148523 -3.375039
3 0.182864 -4.209453 0.314109 0.648528
4 -4.000000 0.321928 1.000000 -0.540568
5 -0.222392 -0.029747 0.085730 0.140178
6 0.697437 0.597902 -2.209453 -0.624491
7 0.736966 -0.584962 0.502500 -2.000000
seqlogo.CompletePm demo
Here is a quickstart guide on how to leverage the power of seqlogo.CompletePm
# Setting seed for demonstration purposes
>>> np.random.seed(42)
# Making a fake PWM
>>> random_ppm = np.random.dirichlet(np.ones(4), size=6)
>>> cpm = seqlogo.CompletePM(ppm = random_ppm)
# Pfm was imputed
>>> print(cpm.pfm)
A C G T
0 8 52 23 15
1 7 7 2 83
2 16 21 0 61
3 73 9 8 8
4 17 36 28 17
5 49 7 18 24
# Shows the how the PPM data was formatted
>>> print(cpm.ppm)
A C G T
0 0.082197 0.527252 0.230641 0.159911
1 0.070375 0.070363 0.024826 0.834435
2 0.161962 0.216972 0.003665 0.617401
3 0.735638 0.098290 0.082638 0.083434
4 0.179898 0.368931 0.280463 0.170708
5 0.498510 0.079138 0.182004 0.240349
# Computing the PWM using default background and pseudocounts
>>> print(cpm.pwm)
A C G T
0 -1.604773 1.076564 -0.116281 -0.644662
1 -1.828788 -1.829031 -3.331983 1.738871
2 -0.626276 -0.204418 -6.091862 1.304279
3 1.557068 -1.346815 -1.597049 -1.583223
4 -0.474749 0.561423 0.165882 -0.550396
5 0.995695 -1.659494 -0.457960 -0.056800
# See the consensus sequence
>>> print(cpm.consensus)
CTTACA
# See the Information Content
>>> print(cpm.ic)
0 0.305806
1 1.110856
2 0.637149
3 0.748989
4 0.074286
5 0.268034
dtype: float64
Plot the sequence logo with information content scaling
# Setting seed for demonstration purposes
>>> np.random.seed(42)
# Making a fake PWM
>>> random_ppm = np.random.dirichlet(np.ones(4), size=6)
>>> ppm = seqlogo.Ppm(random_ppm)
>>> seqlogo.seqlogo(ppm, ic_scale = False, format = 'png', size = 'medium')
The above code will produce:

Plot the sequence logo with no information content scaling
# Setting seed for demonstration purposes
>>> np.random.seed(42)
# Making a fake PWM
>>> random_ppm = np.random.dirichlet(np.ones(4), size=6)
>>> ppm = seqlogo.Ppm(random_ppm)
>>> seqlogo.seqlogo(ppm, ic_scale = False, format = 'png', size = 'medium')
The above code will produce:

Documentation
seqlogo exposes 5 classes to the user for handling PM data:
seqlogo.Pm: the base class for all other specialized PM subclassesseqlogo.Pfm: The class used for handling PFM dataseqlogo.Ppm: The class used for handling PPM dataseqlogo.Pwm: The class used for handling PWM dataseqlogo.CompletePm: This final class will take any/all of the other PM subclass data and compute any of the other missing data. That is, if the user only provides aseqlogo.Pfmand passes it toseqlogo.CompletePm, it will solve for the PPM, PWM, consensus sequence, and information content.
Additionally, seqlogo also provides 6 methods for converting PM structures:
seqlogo.pfm2ppm: converts a PFM to a PPMseqlogo.pfm2pwm: converts a PFM to a PWMseqlogo.ppm2pfm: converts a PPM to a PFMseqlogo.ppm2pwm: converts a PPM to a PWMseqlogo.pwm2pfm: converts a PWM to a PFMseqlogo.pwm2ppm: converts a PWM to a PPM
The signatures for each item above are as follows:
Classes
seqlogo.CompletePm(pfm = None, ppm = None, pwm = None, background = None, pseudocount = None,
alphabet_type = 'DNA', alphabet = None, default_pm = 'ppm'):
"""
Creates the CompletePm instance. If the user does not define any `pm_filename_or_array`,
it will be initialized to empty. Will generate all other attributes as soon
as a `pm_filename_or_array` is supplied.
Args:
pfm (str or `numpy.ndarray` or `pandas.DataFrame` or Pm): The user supplied
PFM. If it is a filename, the file will be opened
and parsed. If it is an `numpy.ndarray` or `pandas.DataFrame`,
it will just be assigned. (default: None, skips '#' comment lines)
ppm (str or `numpy.ndarray` or `pandas.DataFrame` or Pm): The user supplied
PPM. If it is a filename, the file will be opened
and parsed. If it is an `numpy.ndarray` or `pandas.DataFrame`,
it will just be assigned. (default: None, skips '#' comment lines)
pwm (str or `numpy.ndarray` or `pandas.DataFrame` or Pm): The user supplied
PWM. If it is a filename, the file will be opened
and parsed. If it is an `numpy.ndarray` or `pandas.DataFrame`,
it will just be assigned. (default: None, skips '#' comment lines)
background (constant or Collection): Offsets used to calculate background letter probabilities (defaults: If
using an Nucleic Acid alphabet: 0.25; if using an Aminio Acid alphabet: Robinson-Robinson Frequencies)
pseudocount (constant): Some constant to offset PPM conversion to PWM to prevent -/+ inf. (defaults to 1e-10)
alphabet_type (str): Desired alphabet to use. Order matters (default: 'DNA')
"DNA" := "ACGT"
"reduced DNA" := "ACGTN-"
"ambig DNA" := "ACGTRYSWKMBDHVN-"
"RNA" := "ACGU"
"reduced RNA" := "ACGUN-"
"ambig RNA" := "ACGURYSWKMBDHVN-"
"AA" : = "ACDEFGHIKLMNPQRSTVWY"
"reduced AA" := "ACDEFGHIKLMNPQRSTVWYX*-"
"ambig AA" := "ACDEFGHIKLMNOPQRSTUVWYBJZX*-"
"custom" := None
(default: 'DNA')
alphabet (str): if 'custom' is selected or a specialize alphabet is desired, this accepts a string (default: None)
default_pm (str): which of the 3 pm's do you want to call '*home*'? (default: 'ppm')
"""
seqlogo.Pm(pm_filename_or_array = None, pm_type = 'ppm', alphabet_type = 'DNA', alphabet = None,
background = None, pseudocount = None):
"""Initializes the Pm
Creates the Pm instance. If the user does not define `pm_filename_or_array`,
it will be initialized to empty. Will generate all other attributes as soon
as a `pm_filename_or_array` is supplied.
Args:
pm_filename_or_array (str or `numpy.ndarray` or `pandas.DataFrame` or Pm): The user supplied
PM. If it is a filename, the file will be opened
and parsed. If it is an `numpy.ndarray` or `pandas.DataFrame`,
it will just be assigned. (default: None, skips '#' comment lines)
alphabet_type (str): Desired alphabet to use. Order matters (default: 'DNA')
"DNA" := "ACGT"
"reduced DNA" := "ACGTN-"
"ambig DNA" := "ACGTRYSWKMBDHVN-"
"RNA" := "ACGU"
"reduced RNA" := "ACGUN-"
"ambig RNA" := "ACGURYSWKMBDHVN-"
"AA" : = "ACDEFGHIKLMNPQRSTVWY"
"reduced AA" := "ACDEFGHIKLMNPQRSTVWYX*-"
"ambig AA" := "ACDEFGHIKLMNOPQRSTUVWYBJZX*-"
"custom" := None
(default: 'DNA')
alphabet (str): if 'custom' is selected or a specialize alphabet is desired, this accepts a string (default: None)
background (constant or Collection): Offsets used to calculate background letter probabilities (defaults: If
using an Nucleic Acid alphabet: 0.25; if using an Aminio Acid alphabet: Robinson-Robinson Frequencies)
pseudocount (constant): Some constant to offset PPM conversion to PWM to prevent -/+ inf. (default: 1e-10)
"""
seqlogo.Pfm(pfm_filename_or_array = None, pm_type = 'pfm', alphabet_type = 'DNA', alphabet = None,
background = None, pseudocount = None):
"""Initializes the Pfm
Creates the Pfm instance. If the user does not define `pfm_filename_or_array`,
it will be initialized to empty. Will generate all other attributes as soon
as a `pfm_filename_or_array` is supplied.
Args:
pfm_filename_or_array (str or `numpy.ndarray` or `pandas.DataFrame` or Pm): The user supplied
PFM. If it is a filename, the file will be opened
and parsed. If it is an `numpy.ndarray` or `pandas.DataFrame`,
it will just be assigned. (default: None, skips '#' comment lines)
alphabet_type (str): Desired alphabet to use. Order matters (default: 'DNA')
"DNA" := "ACGT"
"reduced DNA" := "ACGTN-"
"ambig DNA" := "ACGTRYSWKMBDHVN-"
"RNA" := "ACGU"
"reduced RNA" := "ACGUN-"
"ambig RNA" := "ACGURYSWKMBDHVN-"
"AA" : = "ACDEFGHIKLMNPQRSTVWY"
"reduced AA" := "ACDEFGHIKLMNPQRSTVWYX*-"
"ambig AA" := "ACDEFGHIKLMNOPQRSTUVWYBJZX*-"
"custom" := None
(default: 'DNA')
alphabet (str): if 'custom' is selected or a specialize alphabet is desired, this accepts a string (default: None)
background (constant or Collection): Offsets used to calculate background letter probabilities (defaults: If
using an Nucleic Acid alphabet: 0.25; if using an Aminio Acid alphabet: Robinson-Robinson Frequencies)
pseudocount (constant): Some constant to offset PPM conversion to PWM to prevent -/+ inf. (default: 1e-10)
"""
seqlogo.Ppm(ppm_filename_or_array = None, pm_type = 'ppm', alphabet_type = 'DNA', alphabet = None,
background = None, pseudocount = None):
"""Initializes the Ppm
Creates the Ppm instance. If the user does not define `ppm_filename_or_array`,
it will be initialized to empty. Will generate all other attributes as soon
as a `ppm_filename_or_array` is supplied.
Args:
ppm_filename_or_array (str or `numpy.ndarray` or `pandas.DataFrame` or Pm): The user supplied
PPM. If it is a filename, the file will be opened
and parsed. If it is an `numpy.ndarray` or `pandas.DataFrame`,
it will just be assigned. (default: None, skips '#' comment lines)
alphabet_type (str): Desired alphabet to use. Order matters (default: 'DNA')
"DNA" := "ACGT"
"reduced DNA" := "ACGTN-"
"ambig DNA" := "ACGTRYSWKMBDHVN-"
"RNA" := "ACGU"
"reduced RNA" := "ACGUN-"
"ambig RNA" := "ACGURYSWKMBDHVN-"
"AA" : = "ACDEFGHIKLMNPQRSTVWY"
"reduced AA" := "ACDEFGHIKLMNPQRSTVWYX*-"
"ambig AA" := "ACDEFGHIKLMNOPQRSTUVWYBJZX*-"
"custom" := None
(default: 'DNA')
alphabet (str): if 'custom' is selected or a specialize alphabet is desired, this accepts a string (default: None)
background (constant or Collection): Offsets used to calculate background letter probabilities (defaults: If
using an Nucleic Acid alphabet: 0.25; if using an Aminio Acid alphabet: Robinson-Robinson Frequencies)
pseudocount (constant): Some constant to offset PPM conversion to PWM to prevent -/+ inf. (default: 1e-10)
"""
seqlogo.Pwm(pwm_filename_or_array = None, pm_type = 'pwm', alphabet_type = 'DNA', alphabet = None,
background = None, pseudocount = None):
"""Initializes the Pwm
Creates the Pwm instance. If the user does not define `pwm_filename_or_array`,
it will be initialized to empty. Will generate all other attributes as soon
as a `pwm_filename_or_array` is supplied.
Args:
pwm_filename_or_array (str or `numpy.ndarray` or `pandas.DataFrame` or Pm): The user supplied
PWM. If it is a filename, the file will be opened
and parsed. If it is an `numpy.ndarray` or `pandas.DataFrame`,
it will just be assigned. (default: None, skips '#' comment lines)
alphabet_type (str): Desired alphabet to use. Order matters (default: 'DNA')
"DNA" := "ACGT"
"reduced DNA" := "ACGTN-"
"ambig DNA" := "ACGTRYSWKMBDHVN-"
"RNA" := "ACGU"
"reduced RNA" := "ACGUN-"
"ambig RNA" := "ACGURYSWKMBDHVN-"
"AA" : = "ACDEFGHIKLMNPQRSTVWY"
"reduced AA" := "ACDEFGHIKLMNPQRSTVWYX*-"
"ambig AA" := "ACDEFGHIKLMNOPQRSTUVWYBJZX*-"
"custom" := None
(default: 'DNA')
alphabet (str): if 'custom' is selected or a specialize alphabet is desired, this accepts a string (default: None)
background (constant or Collection): Offsets used to calculate background letter probabilities (defaults: If
using an Nucleic Acid alphabet: 0.25; if using an Aminio Acid alphabet: Robinson-Robinson Frequencies)
pseudocount (constant): Some constant to offset PPM conversion to PWM to prevent -/+ inf. (default: 1e-10)
"""
Conversion Methods
seqlogo.pfm2ppm(pfm):
"""Converts a Pfm to a ppm array
Args:
pfm (Pfm): a fully initialized Pfm
Returns:
(np.array): converted values
"""
seqlogo.pfm2pwm(pfm, background = None, pseudocount = None):
"""Converts a Pfm to a pwm array
Args:
pfm (Pfm): a fully initialized Pfm
background: accounts for relative weights from background. Must be a constant or same number of columns as Pwm (default: None)
pseudocount (const): The number used to offset log-likelihood conversion from probabilites (default: None -> 1e-10)
Returns:
(np.array): converted values
"""
seqlogo.ppm2pfm(ppm):
"""Converts a Ppm to a pfm array
Args:
ppm (Ppm): a fully initialized Ppm
Returns:
(np.array): converted values
"""
seqlogo.ppm2pwm(ppm, background= None, pseudocount = None):
"""Converts a Ppm to a pwm array
Args:
ppm (Ppm): a fully initialized Ppm
background: accounts for relative weights from background. Must be a constant or same number of columns as Pwm (default: None)
pseudocount (const): The number used to offset log-likelihood conversion from probabilites (default: None -> 1e-10)
Returns:
(np.array): converted values
Raises:
ValueError: if the pseudocount isn't a constant or the same length as sequence
"""
seqlogo.pwm2pfm(pwm, background = None, pseudocount = None):
"""Converts a Pwm to a pfm array
Args:
pwm (Pwm): a fully initialized Pwm
background: accounts for relative weights from background. Must be a constant or same number of columns as Pwm (default: None)
pseudocount (const): The number used to offset log-likelihood conversion from probabilites (default: None -> 1e-10)
Returns:
(np.array): converted values
"""
seqlogo.pwm2ppm(pwm, background = None, pseudocount = None):
"""Converts a Pwm to a ppm array
Args:
pwm (Pwm): a fully initialized Pwm
background: accounts for relative weights from background. Must be a constant or same number of columns as Pwm (default: None)
pseudocount (const): The number used to offset log-likelihood conversion from probabilites (default: None -> 1e-10)
Returns:
(np.array): converted values
Raises:
ValueError: if the pseudocount isn't a constant or the same length as sequence
"""
Contributing
Please see our contribution guidelines here
Acknowledgments
- Bembom O (2018). seqlogo: Sequence logos for DNA sequence alignments. R package version 1.48.0.
- Crooks GE, Hon G, Chandonia JM, Brenner SE WebLogo: A sequence logo generator, Genome Research, 14:1188-1190, (2004).
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file seqlogo-5.29.11.tar.gz.
File metadata
- Download URL: seqlogo-5.29.11.tar.gz
- Upload date:
- Size: 29.3 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.11.11
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
db5700e49c4f0d435db2b3e59c8890fb37364ff5ce015d70e92b0a514e8378a5
|
|
| MD5 |
c5f42f718c5a60df22b911be9e1b89b4
|
|
| BLAKE2b-256 |
6ec002f22f60bdf3ea4d2efe2f2d7532af6ca461cf7e09d46ccae23b43448299
|
File details
Details for the file seqlogo-5.29.11-py2.py3-none-any.whl.
File metadata
- Download URL: seqlogo-5.29.11-py2.py3-none-any.whl
- Upload date:
- Size: 20.1 kB
- Tags: Python 2, Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.11.11
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
fc72f3ce00e91960b36c3fd2d5fb66f0359275789287d18126e8e2ee0fa439c1
|
|
| MD5 |
9a7a1894173709d5a760f4212017ada7
|
|
| BLAKE2b-256 |
c99c5ca3551c3862b79d3f43b47607c4684c5f58629da83af534f9bf3da3499b
|







