




This is is the demultiplex pipeline from the Sequana projet
- Overview:
Runs bcl2fastq on raw BCL data and creates plots to ease the QC validation
- Input:
A valid Illumina base calling directory and sample sheet file
- Output:
An HTML report, a set of PNG files and the expected FastQ files
- Status:
production
- Wiki:
- Documentation:
This README file, the Wiki from the github repository (link above) and https://sequana.readthedocs.io
- Citation:
Cokelaer et al, (2017), ‘Sequana’: a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI https://doi:10.21105/joss.00352
Installation
Intall the sequana_demultiplex package as follows:
pip install sequana_demultiplex
Usage
sequana_demultiplex --help sequana_demultiplex --working-directory DATAPATH --bcl-directory bcldata --sample-sheet SampleSheet.csv --merging-strategy merge
The –bcl-directory option indicates where to find your raw data, the sample-sheet expects the SampleSheet to be compatible with IEM software. The –merging-strategy can be set to none or merge. The merge option merges the lanes, which is useful for e.g. NextSeq sequencers.
This creates a directory fastq. You just need to execute the pipeline:
cd demultiplex sh demultiplex.sh # for a local run
These commands launch a snakemake pipeline. If you are familiar with snakemake, you can retrieve the demultiplex.rules and config.yaml files and then execute the pipeline yourself with specific parameters:
snakemake -s demultiplex.rules --cores 4 --stats stats.txt \
--wrapper-prefix https://raw.githubusercontent.com/sequana/sequana-wrappers/"
You may also use sequanix for a graphical interface.
Would you need to merge the lane, please add the –merging-strategy argument followed by merge:
sequana_demultiplex --bcl-directory bcl_data --merging-strategy merge --sample-sheet SampleSheet.csv
Requirements
This pipeline requires the following third-party tool(s):
bcl2fastq 2.20.0
This software has an end-user license agreement (EULA). Given the EULA details of this software, it cannot be distributed according to ` Illumina license <https://support.illumina.com/content/dam/illumina-support/documents/downloads/software/bcl2fastq/bcl2fastq2-v2-20-eula.pdf>`_ Therefore, you should install it yourself. On cluster facility, you may ask to your system administator. For instance:
module load bcl2fastq/2.20.0
For the same reason you cannot find it on community such as bioconda or docker (aug 2020).
So, you will need to download the code yourself. The easiest is to download the RPM from Illumina and accept the agreements. Install it using the RPM if you have a debian-like system:
rpm install file.rpm
If you do not have a debian system, you can look at https://damona.readthedocs.io where we provide a singularity recipes to build an image from your own rpm. Recipes can be found here.
Details
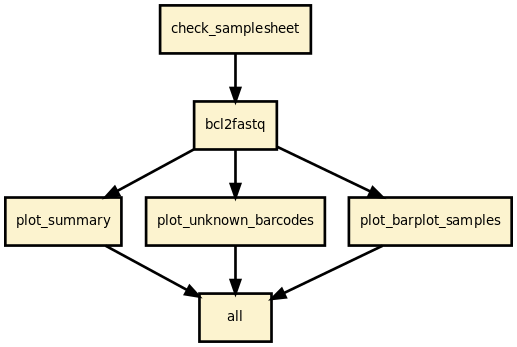
This pipeline runs bcl2fastq 2.20 and creates a set of diagnostics plots to help deciphering common issues such as missing index and sample sheet errors.
Rules and configuration details
Here is the latest documented configuration file to be used with the pipeline. Each rule used in the pipeline may have a section in the configuration file.
Changelog
Version |
Description |
|---|---|
1.6.0 |
|
1.5.2 |
|
1.5.1 |
|
1.5.0 |
|
1.4.0 |
|
1.3.1 |
|
1.3.0 |
|
1.2.1 |
|
1.2.0 |
|
1.1.3 |
|
1.1.2 |
|
1.1.1 |
|
1.1.0 |
|
1.0.5 |
|
1.0.4 |
|
1.0.3 |
|
1.0.2 |
Use ‘sequana samplesheet –check ‘ command instead of deprecated sequana_check_sample_sheet command |
1.0.1 |
change some default behaviour:
|
1.0.0 |
|
0.9.11 |
|
0.9.10 |
|
0.9.9 |
|
0.9.8 |
|
0.9.7 |
|
0.9.6 |
|
0.9.5 |
|
0.9.4 |
|
0.9.3 |
Fix regression bug |
0.9.2 |
remove warning due to relative paths. |
0.9.1 |
Make the merging options compulsory. Users must tell whether they want to merge the lanes or not. This avoid to do the merging or not whereas the inverse was expected. |
0.8.6 |
Uses 64G/biomics queue and 16 cores on a SLURM scheduler |
Contribute & Code of Conduct
To contribute to this project, please take a look at the Contributing Guidelines first. Please note that this project is released with a Code of Conduct. By contributing to this project, you agree to abide by its terms.
1 maintainer
Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file sequana_demultiplex-1.6.0.tar.gz.
File metadata
- Download URL: sequana_demultiplex-1.6.0.tar.gz
- Upload date:
- Size: 44.7 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: poetry/2.0.1 CPython/3.10.14 Linux/6.14.5-100.fc40.x86_64
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
46458ed7348174687612d73b55c16e2ea7d44ad58eacaedfcb42350b513c515b
|
|
| MD5 |
f9d6d8a782b8ae0bacb52666882d8230
|
|
| BLAKE2b-256 |
306e19dc4095811f94ea4cc46f9445d7203dcd1b9867d0a1ab0ffa34fe5f5c52
|
File details
Details for the file sequana_demultiplex-1.6.0-py3-none-any.whl.
File metadata
- Download URL: sequana_demultiplex-1.6.0-py3-none-any.whl
- Upload date:
- Size: 43.1 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: poetry/2.0.1 CPython/3.10.14 Linux/6.14.5-100.fc40.x86_64
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
4a99d04bd3fa9b31d0bbc2788f98eb3f8361d88207510b8ca48b926000b7bb1e
|
|
| MD5 |
0c7324607ae426c663459b8ee00ce990
|
|
| BLAKE2b-256 |
c2656f77042a73d77a3c9d21ad1544b25dd3dfd3bdd67be4b11585905cf54eaf
|









