A multi-sample mapper to map reads onto a reference

Project description




This is the mapper pipeline from the Sequana projet
- Overview:
This is a simple pipeline to map several FastQ files onto a reference using different mappers/aligners
- Input:
A set of FastQ files (illumina, pacbio, etc).
- Output:
A set of BAM files (and/or bigwig) and HTML report
- Status:
Production
- Documentation:
This README file, and https://sequana.readthedocs.io
- Citation:
Cokelaer et al, (2017), ‘Sequana’: a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI https://doi:10.21105/joss.00352
Installation
If you already have all requirements, you can install the packages using pip:
pip install sequana_mapper --upgrade
You will need third-party software such as bwa and samtools. Please see below for details.
Usage
Scan FastQ files in a directory and set up the pipeline (replace DATAPATH and genome.fa with your inputs):
sequana_mapper --input-directory DATAPATH --reference-file genome.fa --aligner-choice bwa sequana_mapper --input-directory DATAPATH --reference-file genome.fa --aligner-choice bwa --do-coverage sequana_mapper --input-directory DATAPATH --reference-file genome.fa --aligner-choice bwa --create-bigwig
For long-read data, use the dedicated presets:
sequana_mapper --input-directory DATAPATH --reference-file genome.fa --pacbio # sets minimap2 -x map-pb sequana_mapper --input-directory DATAPATH --reference-file genome.fa --nanopore # sets minimap2 -x map-ont
For capture-seq projects (feature counting):
sequana_mapper --input-directory DATAPATH --reference-file genome.fa --capture-annotation-file targets.saf
This creates a mapper/ directory with the pipeline and configuration file. Execute the pipeline locally:
cd mapper bash mapper.sh
See .sequana/profile/config.yaml to tune Snakemake behaviour (cores, cluster settings, etc.).
Usage with apptainer
With apptainer, initiate the working directory as follows:
sequana_mapper --input-directory DATAPATH --reference-file genome.fa --use-apptainer
Images are downloaded in the working directory but you can store them in a shared location:
sequana_mapper --input-directory DATAPATH --reference-file genome.fa --use-apptainer --apptainer-prefix ~/.sequana/apptainers
and then:
cd mapper bash mapper.sh
Requirements
This pipeline requires the following executables (install via bioconda/conda):
bwa — short-read aligner (default)
minimap2 — long-read aligner (PacBio / Nanopore)
bowtie2 — alternative short-read aligner
samtools / sambamba — BAM processing
bamtools — BAM statistics
fastqc — raw-read quality control (optional, --do-fastqc)
deeptools — bigwig generation (bamCoverage)
bedtools — genome arithmetic
subread — feature counting (featureCounts, capture-seq only)
mosdepth — fast coverage depth
seqkit — FASTQ statistics
multiqc — aggregated HTML report
sequana_coverage — coverage analysis (prokaryotes)
Install all dependencies at once:
mamba env create -f environment.yml
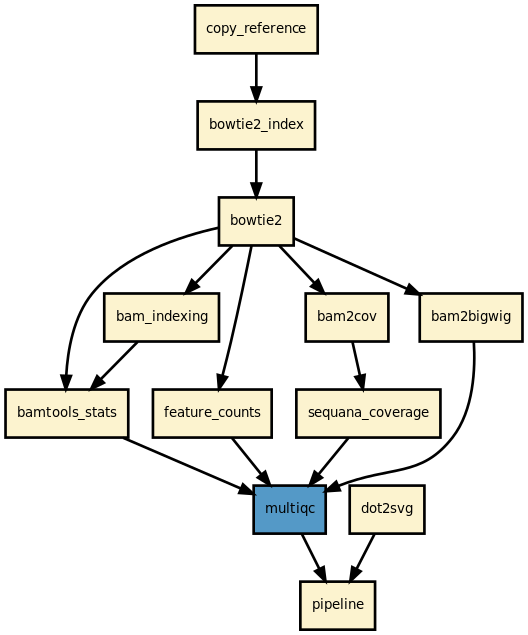
Details
This pipeline maps FastQ files (paired or single-end) in parallel onto a reference genome and produces filtered BAM files, a MultiQC HTML report, and optionally coverage tracks and feature counts.
Aligner choice (--aligner-choice):
bwa (default) — BWA-MEM; index algorithm is auto-selected (is or bwtsw) based on reference size
bwa_split — experimental; splits large FastQs into 1 M-read chunks for parallel BWA jobs, then merges
minimap2 — long-read aligner; use --pacbio (sets -x map-pb) or --nanopore (sets -x map-ont)
bowtie2 — standard short-read aligner
BAM filtering: unmapped reads are removed to minimise file size. Statistics reported by MultiQC (in {sample}/bamtools_stats/) still include both mapped and unmapped read counts.
Optional outputs:
--do-coverage — runs sequana_coverage for depth-of-coverage analysis (prokaryotes)
--do-fastqc — runs fastqc on the raw input reads (included in the MultiQC report)
--create-bigwig — generates bigwig files via bamCoverage (deeptools)
--capture-annotation-file — enables featureCounts for capture-seq efficiency metrics
Rules and configuration details
Here is the latest documented configuration file to be used with the pipeline. Each rule used in the pipeline may have a section in the configuration file.
Changelog
Version |
Description |
|---|---|
1.5.0 |
|
1.4.1 |
|
1.4.0 |
|
1.3.1 |
|
1.3.0 |
|
1.2.1 |
|
1.2.0 |
|
1.1.0 |
|
1.0.0 |
|
0.12.0 |
|
0.11.1 |
|
0.11.0 |
|
0.10.1 |
|
0.10.0 |
|
0.9.0 |
|
0.8.13 |
|
0.8.12 |
|
0.8.11 |
|
0.8.10 |
|
0.8.9 |
|
0.8.8 |
|
0.8.7 |
|
0.8.6 |
|
0.8.5 |
|
0.8.4 |
|
0.8.3 |
|
0.8.2 |
|
0.8.1 |
|
0.8.0 |
First release. |
Contribute & Code of Conduct
To contribute to this project, please take a look at the Contributing Guidelines first. Please note that this project is released with a Code of Conduct. By contributing to this project, you agree to abide by its terms.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file sequana_mapper-1.5.0.tar.gz.
File metadata
- Download URL: sequana_mapper-1.5.0.tar.gz
- Upload date:
- Size: 74.2 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
68265dc658d35ff4e352b32d3dcec9d6e56d3694dc4c435d7cb9869dc613f133
|
|
| MD5 |
a3612cea882e14d8fc942ad21f2bf2f3
|
|
| BLAKE2b-256 |
8ee7622f772a882e5af9dd1a80ffde77e5c8188cc0e2cd252f94d91b8b942de2
|
File details
Details for the file sequana_mapper-1.5.0-py3-none-any.whl.
File metadata
- Download URL: sequana_mapper-1.5.0-py3-none-any.whl
- Upload date:
- Size: 72.6 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
1055442b43a16fd7db3daccfa5f3f18d08bceefaec35f52cccda8f7fa5f6a928
|
|
| MD5 |
a350bf24b813a1c0bd510395da345f79
|
|
| BLAKE2b-256 |
f681e3165994e16acea376b20e56dbdd24507bc2e1536b0b8fe5f39f3be57e58
|







