A multi-sample identification of ribosomal content

Project description




This is is the ribofinder pipeline from the Sequana project
- Overview:
Simple parallele workflow to detect and report ribosomal content
- Input:
FastQ files
- Output:
HTML reports
- Status:
production
- Citation:
Cokelaer et al, (2017), ‘Sequana’: a Set of Snakemake NGS pipelines, Journal of Open Source Software, 2(16), 352, JOSS DOI doi:10.21105/joss.00352
Installation
Using pip from Python, just install this package:
pip install sequana_ribofinder --upgrade
The –upgrade option is to make sure you’ll get the latest version.
Usage
This pipeline scans input fastq.gz files found in the local directory and identify the proportion of ribosomal content.
For help, please type:
sequana_ribofinder --help
The following command searches for input files in DATAPATH. Then, te user provide a list of rRNA sequences in FastA format in test.fasta. This command creates a directory called ribofinder/ where a snakemake pipeline can:
sequana_ribofinder --input-directory DATAPATH --rRNA-file test.fasta
You will then need to execute the pipeline:
cd ribofinder sh ribofinder.sh # for a local run
This launch a snakemake pipeline. If you are familiar with snakemake, you can retrieve the pipeline itself and its configuration files and then execute the pipeline yourself with specific parameters:
snakemake -s ribofinder.rules -c config.yaml --cores 4 --wrapper-prefix git+file:////home/user/sequana_wrappers
Or use sequanix interface.
Requirements
This pipelines requires the following executable(s):
bowtie2 >= 2.4.0
bwa
sambamba
bedtools
samtools
pigz
The aligner is selectable at the command line with --aligner bowtie2 (default) or --aligner bwa. You only need to install the one(s) you intend to use.
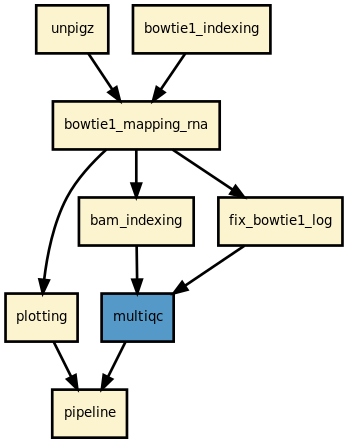
Details
This pipeline runs ribofinder in parallel on the input fastq files. A brief sequana summary report is also produced.
You can start from the reference file and the GFF file. By default we search for the feature called rRNA to be found in the GFF file:
sequana_ribofinder --input-directory . --reference-file genome.fasta --gff-file genome.gff
If the default feature rRNA is not found, no error is raised for now. If you know the expected feature, you can provide it though:
sequana_ribofinder --input-directory . --reference-file genome.fasta --gff-file genome.gff --rRNA-feature gene_rRNA
If you have an existing or custom rRNA file, you can then use it as follows, in which case, no input reference is required:
sequana_ribofinder --input-directory . --rRNA-file ribo.fasta
Rules and configuration details
Here is the latest documented configuration file to be used with the pipeline. Each rule used in the pipeline may have a section in the configuration file.
Changelog
Version |
Description |
|---|---|
1.2.0 |
|
1.1.1 |
|
1.1.0 |
|
1.0.1 |
|
1.0.0 |
|
0.13.0 |
|
0.12.0 |
|
0.11.1 |
|
0.11.0 |
|
0.10.2 |
|
0.10.1 |
|
0.10.0 |
|
0.9.3 |
|
0.9.2 |
First release. |
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file sequana_ribofinder-1.2.0.tar.gz.
File metadata
- Download URL: sequana_ribofinder-1.2.0.tar.gz
- Upload date:
- Size: 36.7 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
8fc0ed00b381d51b620b22ae0b0fc9a6912bfd4e9f10ee36825d67dc0fafe21b
|
|
| MD5 |
5d3d992c49233f6ce28cd96ae680d495
|
|
| BLAKE2b-256 |
97cc1f8252de7a8ec2aa40ab86ea1338ee5a577b819bd567b538f90cb810214f
|
File details
Details for the file sequana_ribofinder-1.2.0-py3-none-any.whl.
File metadata
- Download URL: sequana_ribofinder-1.2.0-py3-none-any.whl
- Upload date:
- Size: 36.0 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.13.12
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
5ca403aa11507eff5740eb91ded914f633ba55063926311ccdab7396227423ad
|
|
| MD5 |
649ddc4398a4f70a50cde52882d9af16
|
|
| BLAKE2b-256 |
a7ee3dc8d97492c200af9606230d4350481f464b42a301544ab9cd42f9311a8d
|







