A tool for diagnosing spinal muscular atrophy (SMA) using exome or genome sequencing data

Project description
SMA Finder
SMA Finder is a tool for diagnosing spinal muscular atrophy (SMA) based on Illumina exome, genome, or targeted sequencing data.
It takes a reference sequence (FASTA) and 1 or more alignment files (CRAM or BAM) as input, evaluates reads at the
c.840 position of SMN1 and SMN2 to detect the most common molecular causes of SMA, and then reports whether it found a complete loss of SMN1.
It has been tested and confirmed to be highly accurate on short read data aligned to GRCh37, GRCh38, or T2T using the BWA aligner.
Limitations:
- does not report SMA carrier status or SMN1/SMN2 copy numbers
- does not detect the ~5% of cases caused by SMN1 loss-of-function mutations that do not involve the c.840 position
- requires at least 14 reads to overlap the c.840 position in SMN1 plus SMN2 in order to make a call
- was developed and tested on Illumina short read sequencing data generated from whole blood DNA and aligned using the BWA aligner. Performance on data from other sequencing technologies, sample types, and alignment pipelines is unknown.
Install
python3 -m pip install sma-finder
Example
Example command:
sma_finder --verbose --hg38-reference-fasta /ref/hg38.fa sample1.cram
Command output:
Input args:
--hg38-reference-fasta: /ref/hg38.fa
--output-tsv: sample1.sma_finder_results.tsv
CRAMS or BAMS: sample1.cram
---
Output row #1:
filename_prefix sample1
file_type cram
genome_version hg38
sample_id s1
sma_status has SMA
confidence_score 168
c840_reads_with_smn1_base_C 0
c840_total_reads 174
Wrote 1 rows to sample1.sma_finder_results.tsv
Usage
Usage help text:
sma_finder --help
usage: sma_finder.py [-h] [--hg37-reference-fasta HG37_REFERENCE_FASTA]
[--hg38-reference-fasta HG38_REFERENCE_FASTA]
[--t2t-reference-fasta T2T_REFERENCE_FASTA]
[-o OUTPUT_TSV] [-v]
cram_or_bam_path [cram_or_bam_path ...]
positional arguments:
cram_or_bam_path One or more CRAM or BAM file paths
optional arguments:
-h, --help show this help message and exit
--hg37-reference-fasta HG37_REFERENCE_FASTA
HG37 reference genome FASTA path. This should be
specified if the input bam or cram is aligned to HG37.
--hg38-reference-fasta HG38_REFERENCE_FASTA
HG38 reference genome FASTA path. This should be
specified if the input bam or cram is aligned to HG38.
--t2t-reference-fasta T2T_REFERENCE_FASTA
T2T reference genome FASTA path. This should be
specified if the input bam or cram is aligned to the
CHM13 telomere-to-telomere benchmark.
-o OUTPUT_TSV, --output-tsv OUTPUT_TSV
Optional output tsv file path
-v, --verbose Whether to print extra details during the run
Output
The output .tsv contains one row per input CRAM or BAM file and has the following columns:
| filename_prefix | CRAM or BAM filename prefix. If the input file is /path/sample1.cram this would be "sample1". |
| file_type | "cram" or "bam" |
| genome_version | "hg37", "hg38", or "t2t" |
| sample_id | sample id from the CRAM or BAM file header (parsed from the read group metadata) |
| sma_status | possible values are: "has SMA" "does not have SMA" "not enough coverage at SMN c.840 position" |
| confidence_score | PHRED-scaled integer score measuring the level of confidence that the sma_status is correct. The bigger the score, the higher the confidence. It is calculated in a similar way to the PL field in GATK HaplotypeCaller genotypes. |
| c840_reads_with_smn1_base_C | number of reads that have a 'C' nucleotide at the c.840 position in SMN1 plus SMN2 |
| c840_total_reads | total number of reads overlapping the c.840 position in SMN1 plus SMN2 |
Combining results from multiple samples
After running SMA Finder on many samples, it often useful to combine the per-sample output tables into a single table. One way to do this is with the following shell command:
combined_table_filename=combined_results.tsv
head -n 1 $(ls *.tsv | head -n 1) > ${combined_table_filename} # get table header from the 1st table
for i in *.tsv; do
tail -n +2 $i >> ${combined_table_filename} # concatenate all tables
done
Plotting combined results
A scatter plot summarizing read counts from many samples can be generated using the plot_SMN1_SMN2_scatter command:
python3 plot_SMN1_SMN2_scatter.py --format svg --format png ${combined_table_filename}
It generates plots like this one which is based on a neuromuscular cohort with 16,626 exomes:
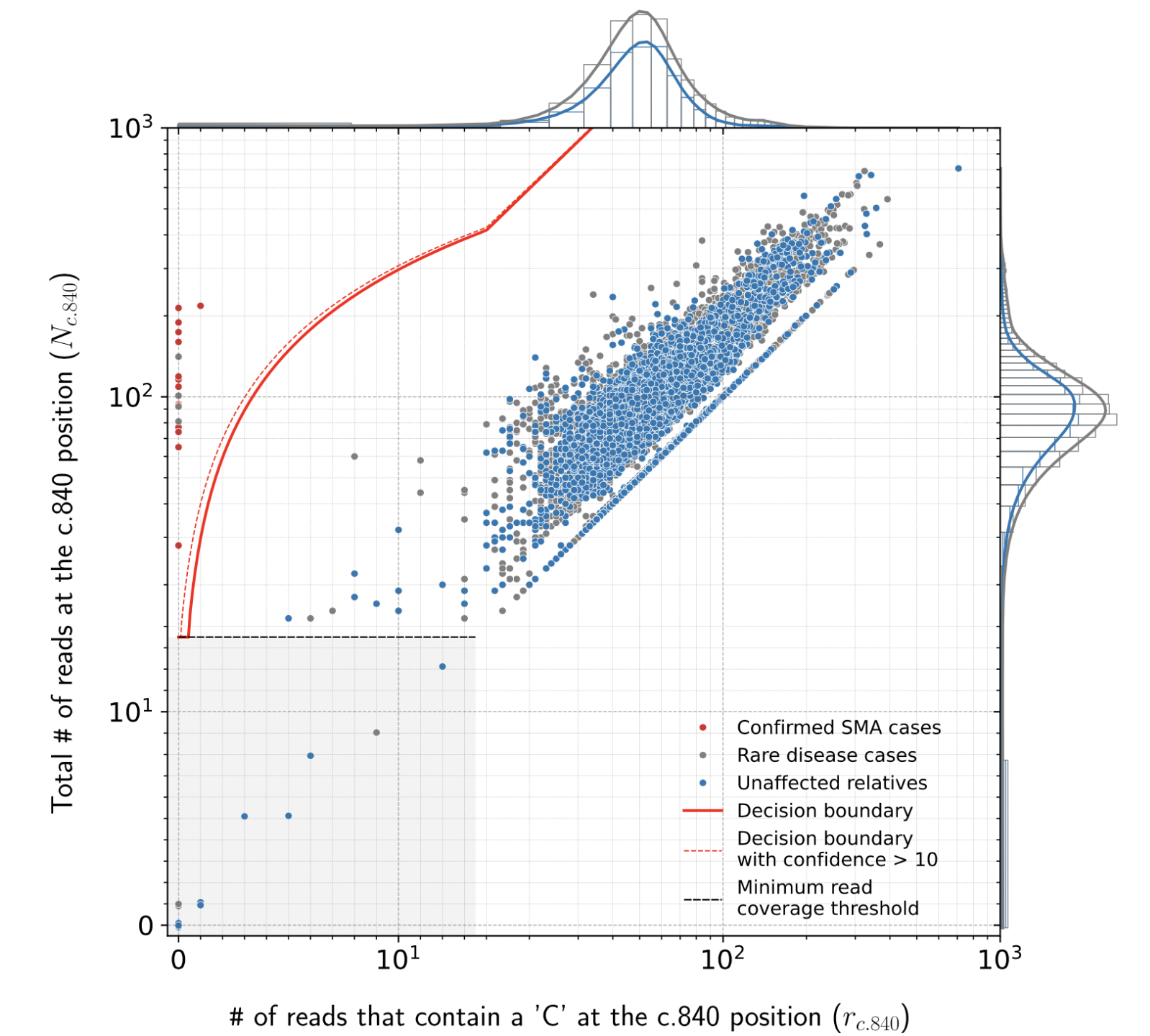
Details
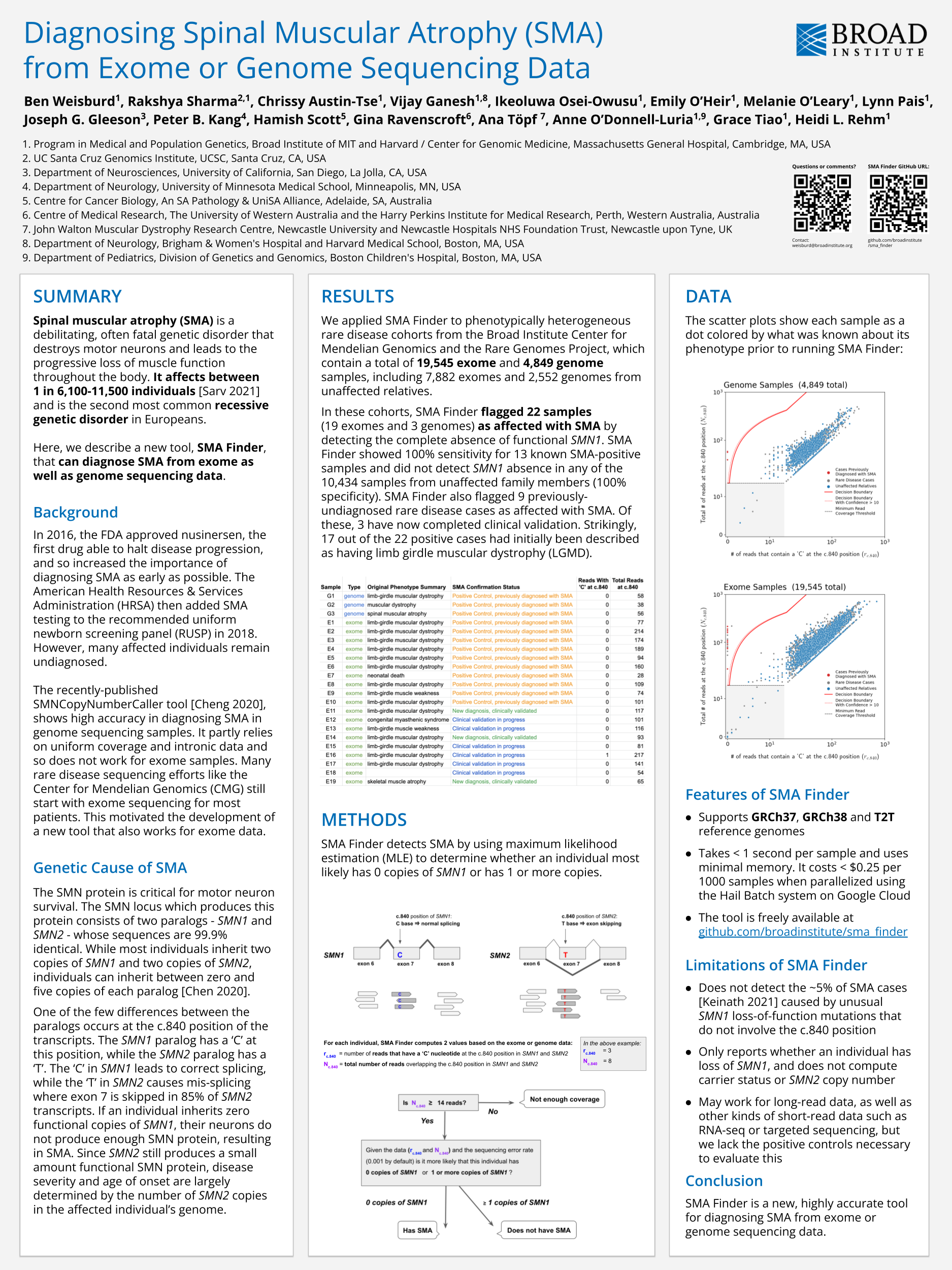
This poster on SMA Finder was presented at the SVAR22 conference:
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file sma_finder-1.4.4.tar.gz.
File metadata
- Download URL: sma_finder-1.4.4.tar.gz
- Upload date:
- Size: 13.6 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.2 CPython/3.9.17
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
e0fdad3e594d1d909fc1f1be91a17464bb64dd0f86096da4b2bb66c4fc92f963
|
|
| MD5 |
c13d70376982e9f431ebd701655f8b69
|
|
| BLAKE2b-256 |
63594b9cb498a264ca79ffe6ac60a3ef21fc83976e6782c5ae184e7341c8ebb7
|
File details
Details for the file sma_finder-1.4.4-py3-none-any.whl.
File metadata
- Download URL: sma_finder-1.4.4-py3-none-any.whl
- Upload date:
- Size: 14.2 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.2 CPython/3.9.17
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
f3f7eb50a71921560194e16c1a8434078a6b9eef5bff8456bb13475b6e05e18f
|
|
| MD5 |
18142d8947d47122328246e7776f1413
|
|
| BLAKE2b-256 |
630f46dfd349199673b74b45708c838729b7db3a6dee53fcdad8d7b219527dfd
|







