A libray for processing sequencing data for SAturated Transposon Analysis in Yeast (SATAY)



Project description
SATAY and Transposonmapper
This workflow is created for processing sequencing data for SAturated Transposon Analysis in Yeast (SATAY) for Saccharomyces Cerevisiae. It performs the steps from raw sequencing data until the transposon mapping that outputs files containing all insertion sites combined with the number of reads.
For more information regarding SATAY, see the satay user website created by the Kornmann-lab. For more extensive documentation, see our JupyterBook.
The workflow requires input sequencing data in fastq format. It can perform the following tasks:
sequence trimming
quality checking raw and trimmed fastq files
sequence alignment with reference genome (S288C Cerevisiae genome)
quality checking bam files, indexing and sorting
transposon mapping
The output files indicate the location of transposon insertions and the number of reads at those locations. This is presented in both .bed and .wig format. Also a list of genes is generated where the number and distribution of insertions and reads is presented per (essential) gene.
Badges |
|
|---|---|
fair-software.nl recommendations |
|
1. Code repository |
|
2. License |
|
3. Community Registry |
|
4. Enable Citation |
|
5. Checklists |
|
Code quality checks |
|
Continuous integration |
|
Documentation |
|
Code Quality |
|













Documentation for users
PyPI package
For users that only require post processing analysis of the data (the bam file was already analyzed), do use the default installation. For example pysam won’t be installed, hence Linux is not required.
pip install transposonmapperFor users that require the whole processing pipeline, do use:
pip install transposonmapper[linux]For more extensive documentation, see our JupyterBook.
SATAY pipeline
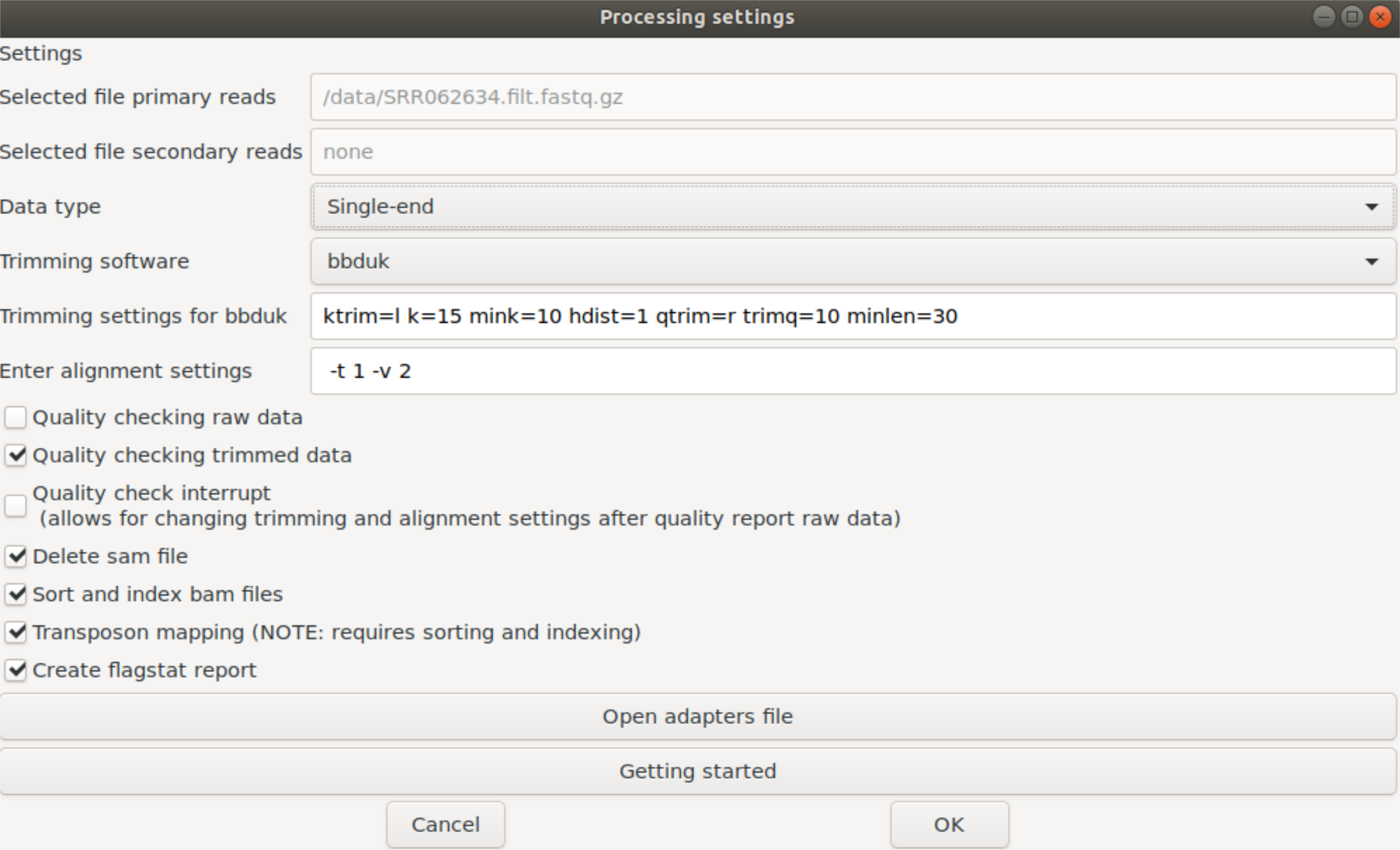
We provide two methods to run the SATAY pipeline, either with a Docker container (recommended) or a Linux system. The workflow relies on the following libraries:
FASTQC v0.11.9 or later
BBMap v38.87 or later
Trimmomatic v0.39 or later
BWA v0.7.17 or later
SAMTools v1.10 or later
BCFTools v1.10.2-3 or later
Sambamba v0.7.1 or later
These libraries are called as a processing pipeline by the script satay.sh, which generates a GUI.
Preprocessing steps
Before inputting the data into the satay pipeline, it is necessary to preprocess the data that comes from the sequencing company.
The pipeline does not process each digestion separately and therefore any pre-processing and trimming of the restriction sites should be done prior to running the pipeline.
What we do if the sequencing data is paired end:
If the data is paired end, only one of the pairs will map to the transposon insertion site (the end that has been sequenced from the sequencing primer), while the other end will map back to a location arbitrarily far upstream or downstream of the insertion site (depends on where the restriction site is).
Preprocessing steps prior to use the satay pipeline:
- Convert the data to single end by:
Extracting the forward reads, which are the reads that contain the sequencing primer, as it is (harsh filtering) or allowing some mismatches in the sequencing primer, due to likely sequencing errors (gentle filtering).
- Remove the sequence downstream the first restriction site for NiaIII and DpnII to avoid having chimeras sequences in our data, that have poor alignment.
Discard reads bellow 50bp after trimming of the restriction site to ensure a decent confidence alignment score for that read.
Docker
For a full installation and user guide for Docker containers, see our documentation.
The Docker image is hosted at leilaicruz/satay.
Prerequisites:
Windows, macOS, Linux
Docker
Xserver (for displaying the GUI)
To build the image locally in your computer, from DockerHub:
create an account in DockerHub
Pull the image
docker pull leilaicruz/satay:latestVerify the image is in your computer
docker imagesMove to where you have the Dockerfile and build the image
docker build . -t leilaicruz/satay:latestMove to the location where you have the data you would like to mount to the container, to use $(pwd) in the command bellow (simplest option), otherwise indicate the absolute path from your computer you would like to be loaded.
To run the docker container, use the commands for your Operating System:
# For Windows (and WSL):
docker run --rm -it -e DISPLAY=host.docker.internal:0 -v /$(pwd):/data leilaicruz/satay:latest
# For macOS
docker run --rm -it -e DISPLAY=docker.for.mac.host.internal:0 -v $(pwd):/data leilaicruz/satay
# For Linux
docker run --rm -it --net=host -e DISPLAY=:0 -v $(pwd):/data leilaicruz/satayThe flag -e enables viewing of the GUI outside the container via the Xserver
The flag -v mounts the current directory (pwd) on the host system to the data/ folder inside the container
Troubleshooting
If an error regarding the connection pops up:
Gtk-WARNING **: cannot open display: :0There is a solution in Linux is typing the following command in the terminal : xhost +
Linux system
Prerequisites:
Anaconda
Python 3.7, 3.8
We recommend installing all dependencies in a conda environment:
git clone https://github.com/SATAY-LL/Transposonmapper.git satay
cd satay
conda env create --file conda/environment-linux.yml
conda activate satay-linuxTo start the GUI, simply run
bash satay.shDocumentation for developers
Installation
To install transposonmapper, do:
git clone https://github.com/SATAY-LL/Transposonmapper.git
cd transposonmapper
conda env create --file conda/environment-dev.yml
conda activate satay-dev
pip install -e .[dev]Run tests (including coverage) with:
pytestDocker image
For more information go to our [Jupyter Book](https://satay-ll.github.io/Transposonmapper/03-docker-doc/03-Docker-Developers.html)
Contributing
If you want to contribute to the development of transposonmapper and the SATAY pipeline, have a look at the contribution guidelines.
Contributors
This software is part of the research effort of the LaanLab, Department of BioNanoScience, Delft University of Technology
Leila Iñigo de la Cruz
Gregory van Beek
Maurits Kok
License
Copyright (c) 2020, Technische Universiteit Delft
Licensed under the Apache License, Version 2.0 (the “License”). The 2.0 version of the Apache License, approved by the ASF in 2004, helps us achieve our goal of providing reliable and long-lived software products through collaborative open source software development.
Unless required by applicable law or agreed to in writing, software distributed under the License is distributed on an “AS IS” BASIS, WITHOUT WARRANTIES OR CONDITIONS OF ANY KIND, either express or implied. See the License for the specific language governing permissions and limitations under the License.


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file transposonmapper-1.1.5.tar.gz.
File metadata
- Download URL: transposonmapper-1.1.5.tar.gz
- Upload date:
- Size: 5.8 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.8.0 pkginfo/1.8.2 readme-renderer/34.0 requests/2.27.1 requests-toolbelt/0.9.1 urllib3/1.26.9 tqdm/4.63.0 importlib-metadata/4.11.3 keyring/23.5.0 rfc3986/2.0.0 colorama/0.4.4 CPython/3.9.11
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d10a628d56f2a1f8be4f3cdcea37b8cd8d1654d0b80219a27bc9dbe9ba82aa3e
|
|
| MD5 |
ebcf88fe5d529fd7b3fa885dba617ea9
|
|
| BLAKE2b-256 |
450038e56c7bb2e96dd1122d4f2855a8e53f593cdf98592d2eb4c160602bd699
|
File details
Details for the file transposonmapper-1.1.5-py3-none-any.whl.
File metadata
- Download URL: transposonmapper-1.1.5-py3-none-any.whl
- Upload date:
- Size: 6.0 MB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.8.0 pkginfo/1.8.2 readme-renderer/34.0 requests/2.27.1 requests-toolbelt/0.9.1 urllib3/1.26.9 tqdm/4.63.0 importlib-metadata/4.11.3 keyring/23.5.0 rfc3986/2.0.0 colorama/0.4.4 CPython/3.9.11
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
d36b782c8930f5c70e66451bc7ce168489dc6bb3453665df0ab7721ab1110097
|
|
| MD5 |
01402f8b5682b777123b88b66edd9d66
|
|
| BLAKE2b-256 |
7e6e34b45648d0e051a889f381a1e019c4fcb0e923da99472785fd1d7daacbc0
|







