Short Tandem Repeat (STR) genotyper

Project description
TREDPARSE: HLI Short Tandem Repeat (STR) caller


| Author | Haibao Tang (tanghaibao) |
| Smriti Ramakrishnan (smr18) | |
| htang@humanlongevity.com | |
| License | See included LICENSE |
Description
Process a list of TRED (trinucleotide repeats disease) loci, and infer the most likely genotype.
Installation
Make sure your Python version ≥ 3.6 (tested in ubuntu, Python 2 no longer supported):
pip install --user -U git+git://github.com/humanlongevity/tredparse.git
For accessing BAMs that are located on S3, please refer to
docker/tredparse.dockerfile for installation of SAMTOOLS/pysam with S3
support.
Or, you can simply build and use the docker image:
docker pull humanlongevity/tredparse
docker run -v `pwd`:`pwd` -w `pwd` humanlongevity/tredparse \
tred.py --tred HD test.bam
Example
First specify the input bam paths and sample keys in a CSV file, like
tests/samples.csv. This file is comma separated:
#SampleKey,BAM,TRED
t001,tests/t001.bam,HD
t002,tests/t002.bam,DM1
If third column is omitted, then all 30 TREDs are scanned. For example:
#SampleKey,BAM
t001,tests/t001.bam
t002,tests/t002.bam
Please also note that the BAM path can start with http:// or s3://, provided
that the corresponding BAM index can be found.
Run tred.py on sample CSV file and generate TSV file with the
genotype:
tred.py tests/samples.csv --workdir work
Highlight the potential risk individuals:
tredreport.py work/*.json --tsv work.tsv
The inferred "at-risk" individuals show up in results:
[DM1] - Myotonic dystrophy 1
rep=CAG inherit=AD cutoff=50 n_risk=1 n_carrier=0 loc=chr19:45770205-45770264
SampleKey inferredGender Calls DM1.FR DM1.PR DM1.RR DM1.PP
t002 Unknown 5|62 5|24 ...|1;39|1;40|1;42|1;43|1;46|2 49|3;50|8 1
[HD] - Huntington disease
rep=CAG inherit=AD cutoff=40 n_risk=1 n_carrier=0 loc=chr4:3074877-3074933
SampleKey inferredGender Calls HD.FR HD.PR HD.RR HD.PP
t001 Unknown 15|41 15|4 ...|1;21|1;24|2;29|1;34|1;41|1 1
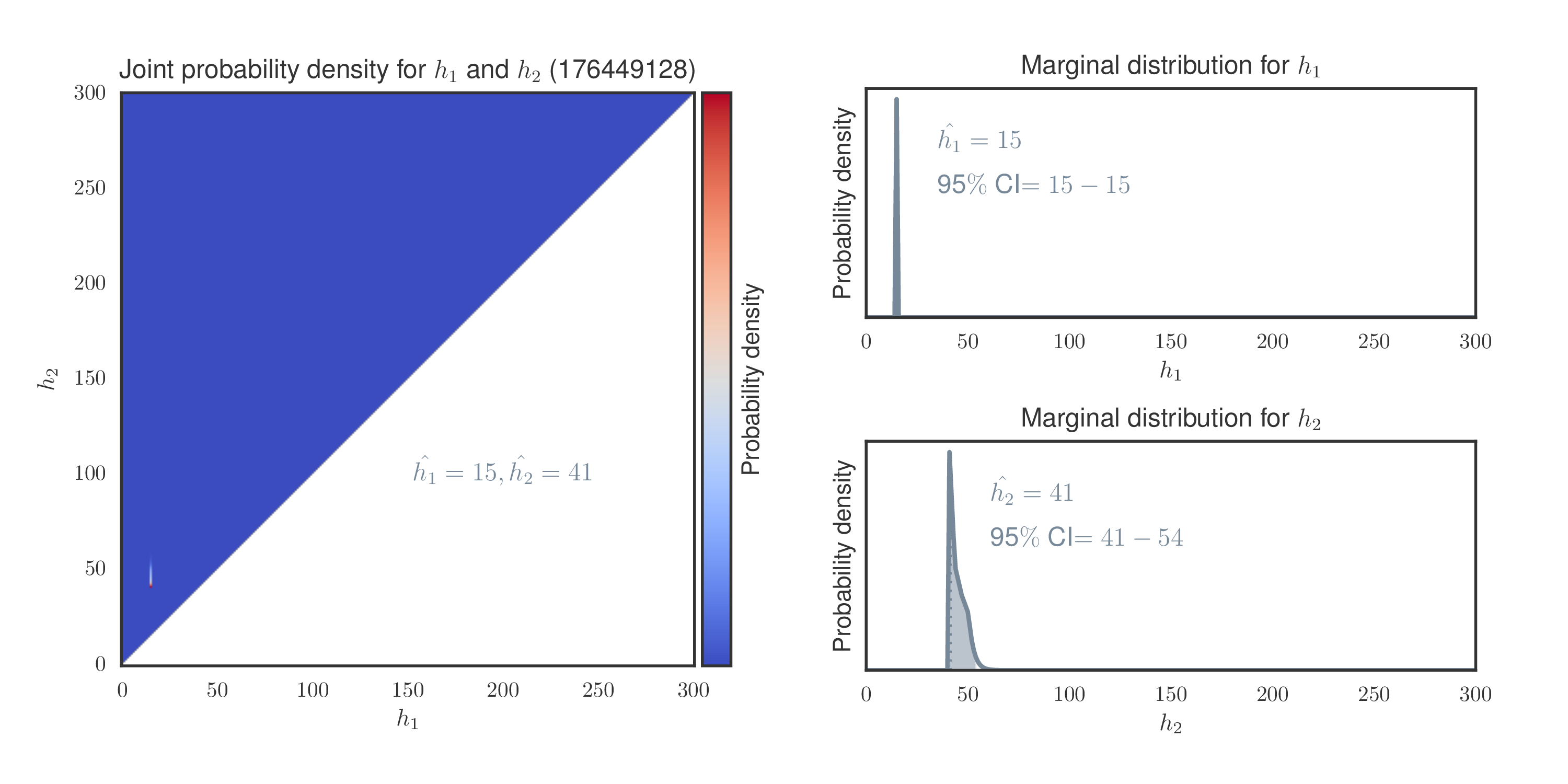
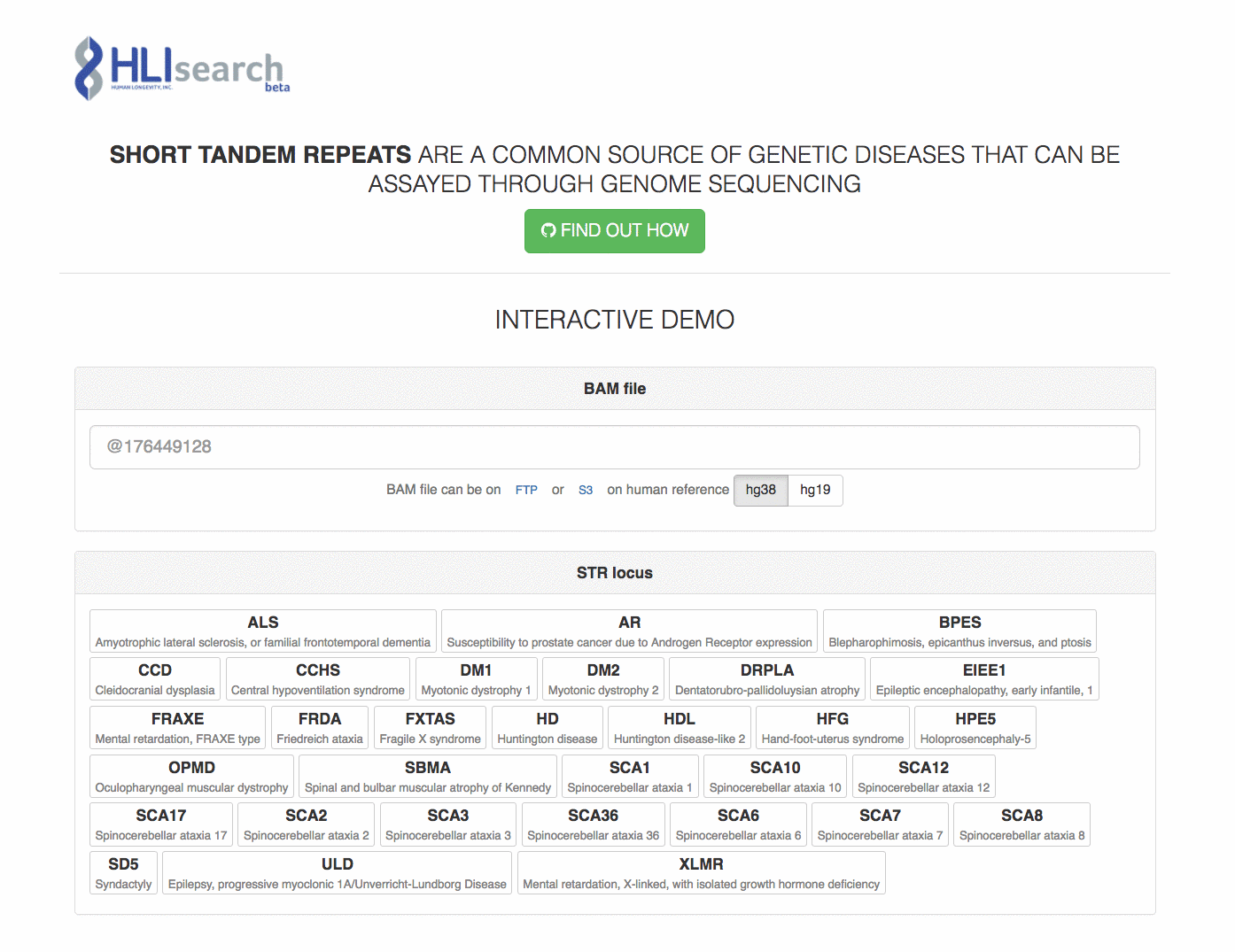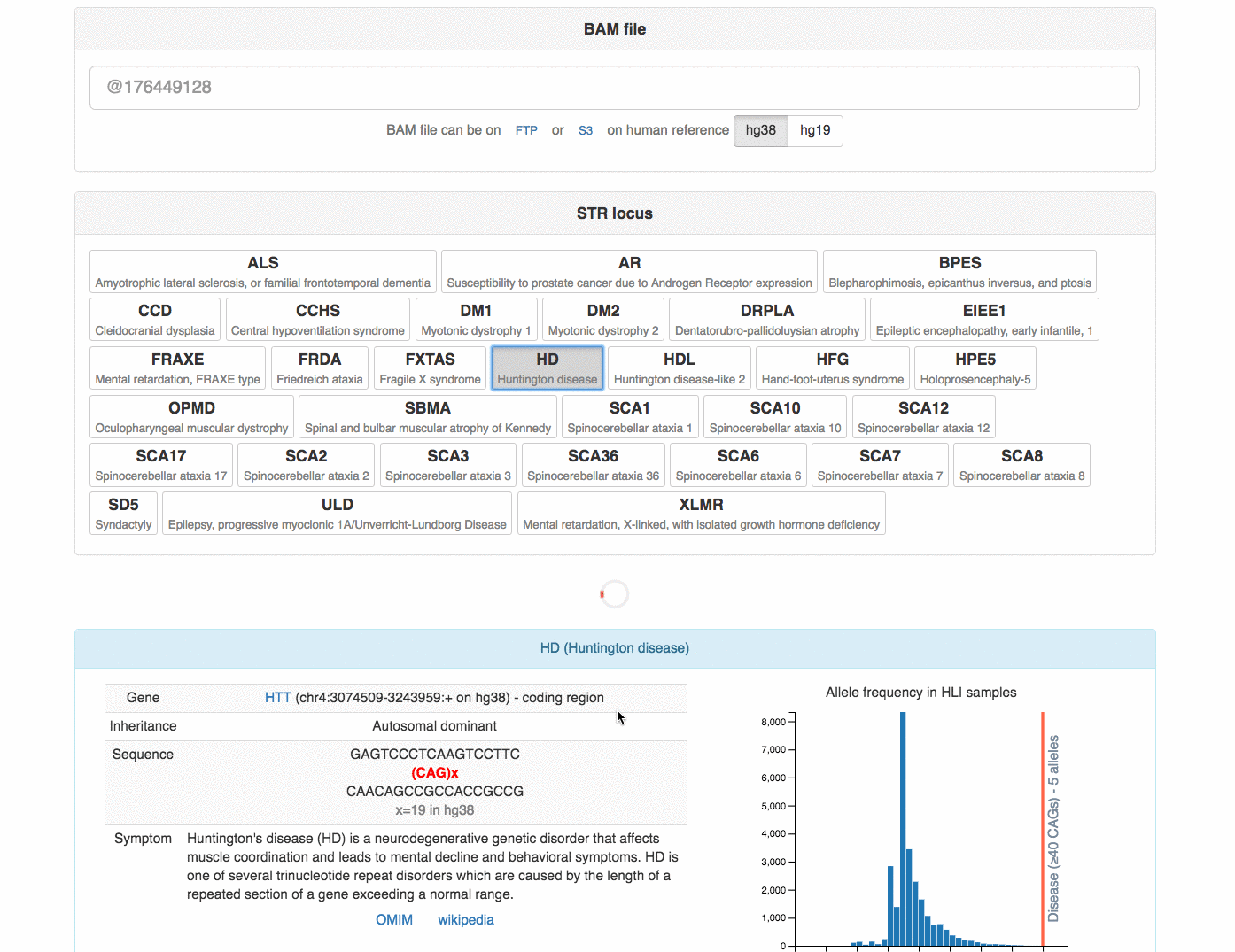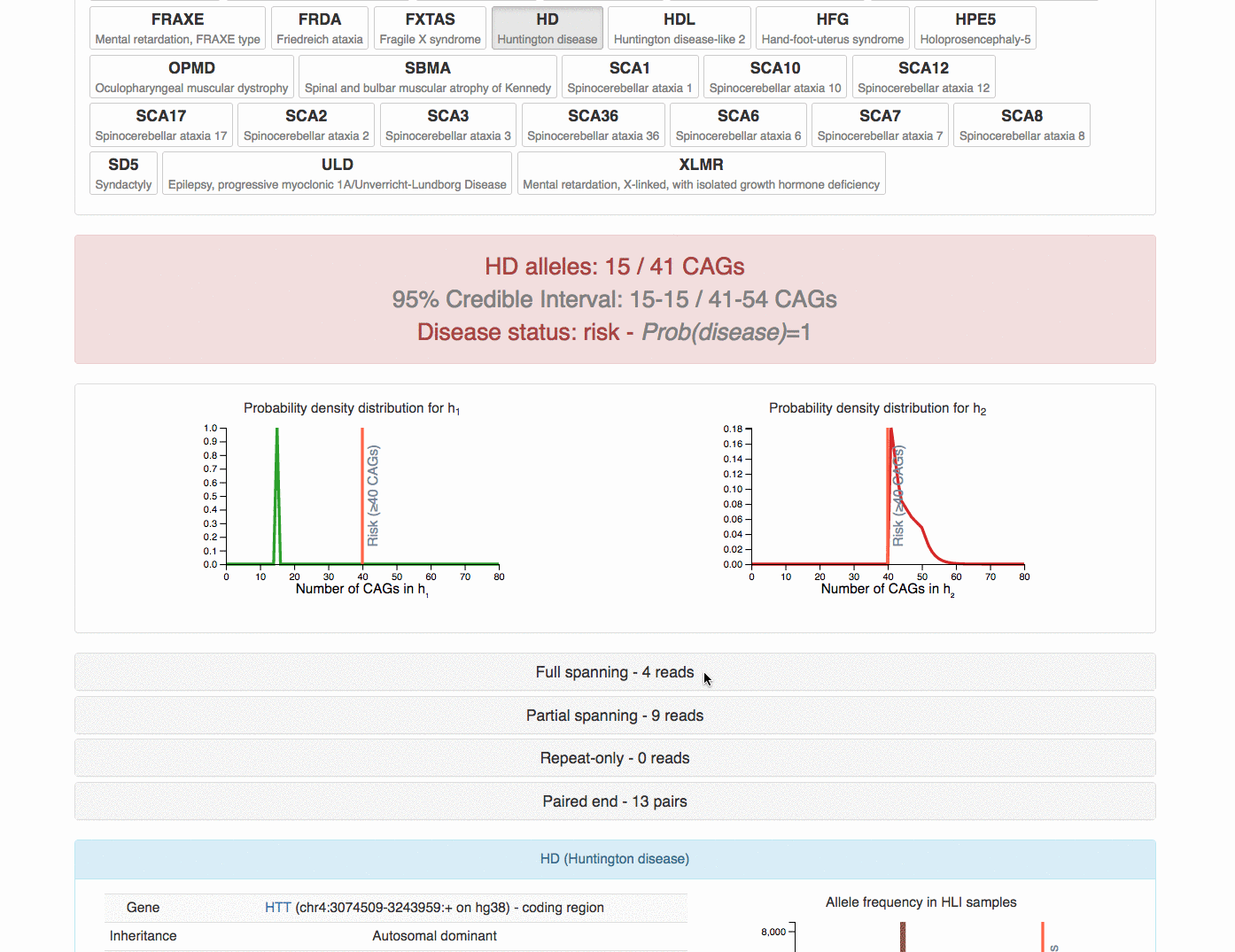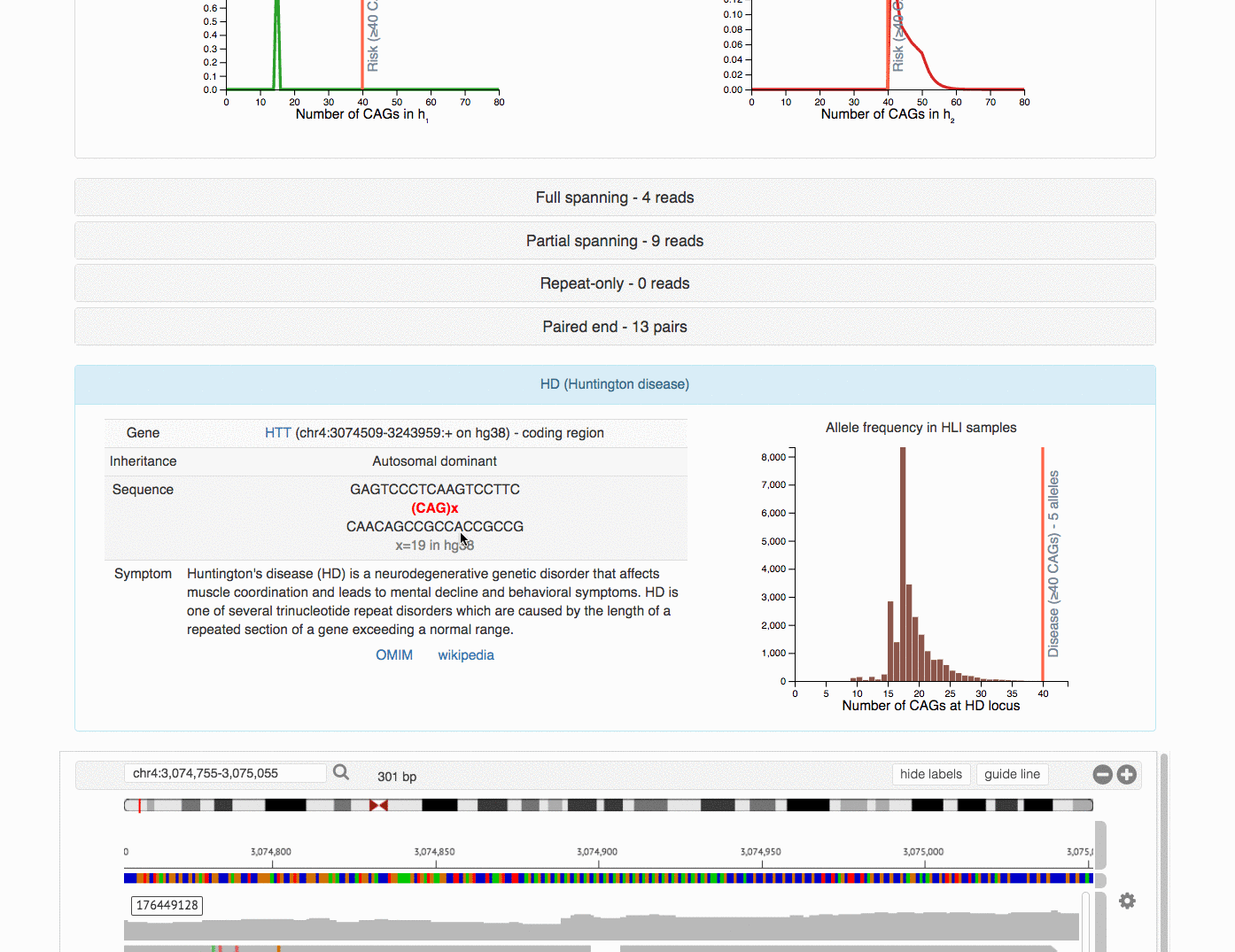
One particular individual t001 appears to have 15/41 call (one allele at 15 CAGs
and the other at 41 CAGs) at Huntington disease locus (HD). Since the risk cutoff
is 40, we have inferred it to be at-risk.
A .report.txt file will also be generated that contains a summary of
number of people affected by over-expanded TREDs as well as population allele
frequency.
To better understand the uncertainties in the prediction, we can plot the
likelihood surface based on the model. Using the same example as above at the
Huntington disease case, we can run a command on the JSON output, with option
--tred HD to specify the locus.
tredplot.py likelihood work/t001.json --tred HD
This generates the following plot:
Server demo
The server/client allows tredparse to be run as a service, also showing the
detailed debug information for the detailed computation.
Install meteor if you don't have it yet.
curl https://install.meteor.com/ | sh
Then build the docker image to run the command, then run the server.
cd docker
make build
cd ../server
meteor npm install
meteor


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file tredparse-1.1.12.tar.gz.
File metadata
- Download URL: tredparse-1.1.12.tar.gz
- Upload date:
- Size: 360.1 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.6.0 importlib_metadata/4.8.2 pkginfo/1.8.1 requests/2.26.0 requests-toolbelt/0.9.1 tqdm/4.62.3 CPython/3.9.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
ff6363365a76952106101fa5d27ccae09ee960ed6d9ea4c6437a0139cf9081b6
|
|
| MD5 |
4fe006975e38e0b6b74cc5f47dad9068
|
|
| BLAKE2b-256 |
3332abbebafed6afbee2bb41e4e0f8be78a4ec2a391dcfef86121acee06b435e
|







