Motif Detection in Multiple Alignment Files: CLI tool to process and analyze MAF files ( Multiple alignment format )

Project description
MAFin: Motif Detection in Multiple Alignment Files
Description
This CLI tool allows you to search MAF (Multiple Alignment Format) files for specific motifs using regular expression patterns, K-mers, or Position Weight Matrices (PWMs). It supports searching within reference genomes or across all genomes and can handle both strands if specified. The tool is optimized for performance with support for parallel processing.
Features
- Motif Search Options: Search using regex patterns, K-mers, or PWMs (JASPAR format).
- Flexible Genome Selection: Choose to search within the reference genome or across all genomes.
- Strand Orientation: Optionally search both strands by considering reverse complements.
- Parallel Processing: Utilize multiple processes to speed up the search.
- Output Formats: By default BED format ( tab separated ) if --detailed_report is specified CSV and JSON analytical report also.
Installation
Prerequisites
- Python 3.6 or higher
pippackage manager- Biopython, pyahocorasick ( installed via requirements.txt )
Using Virtual Environment
-
Clone the Repository
git clone https://github.com/Georgakopoulos-Soares-lab/MAFin cd MAFin
-
Install package inside conda environment
conda create -n mafin_env python=3.8 conda init && conda activate mafin_env pip install -e .
or through PyPI without cloning
pip install MAFin
Usage
MAFin path/to/file.maf \
--regexes "CTGCCCGCA" "AGT" \
--search_in reference \
--reverse_complement no \
--processes 4 \
--detailed_report
Command-Line Arguments
maf_file: (Required) Path to the uncompressed MAF file.
--genome_ids: Path to the genome IDs file (optional).
--search_in: Genome in which to search motifs. Default is 'reference'.
--reverse_complement: Search motifs on both strands if 'yes'. Choices are 'yes' or 'no'. Default is 'no'.
--pvalue_threshold: P-value threshold for PWM matches. Default is 1e-4.
--processes: Number of parallel processes to use. Default is 1.
--background_frequencies: Background nucleotide frequencies for A, C, G, T. They must sum to 1. (Specify as four space-separated floats: A_FREQ C_FREQ G_FREQ T_FREQ)
--purge_results_dir: Purge the 'tmp' directory before running.
--verbose, -v: Enable verbose logging to debug.log.
--detailed_report, -d: Generate merged JSON and CSV output in the current directory.
Search Types (Mutually Exclusive)
--regexes: Regex patterns to search for in the MAF file.
--kmers: Path to the K-mers file (one per line).
--jaspar_file: Path to the PWM file in JASPAR format.
Note: You must specify exactly one of the search types.
Example Usage
Searching with Regex Patterns
MAFin path/to/file.maf \
--regexes "CTG[CC]+CGCA" "AGT" \
--search_in all \
--verbose \
--reverse_complement yes \
--processes 8 \
--purge_results_dir
Searching with K-mers
MAFin path/to/file.maf \
--kmers data/kmers.txt \
--search_in reference \
--processes 2 \
--detailed_report
Searching with PWM (JASPAR format)
MAFin path/to/file.maf \
--jaspar_file data/motif.jaspar \
--search_in all \
--pvalue_threshold 1e-5 \
--background_frequencies 0.25 0.15 0.35 0.25 \
--processes 4
Example Output
BED Output
BED Format (.tsv) with columns chromosome, start , end , motif , conservation , strand
chr14 10685 10701 ATGCATGCATGCATGC 99.11 +
chr24 19318 19334 TTTTTTTTTTTTTTTT 90.18 +
chr24 19458 19474 ATGCATGCATGCATGC 99.11 +
chr29 25671 25687 AAAATTTTGGGGCCCC 100.0 +
chr29 26421 26437 TTTTTTTTTTTTTTTT 85.71 +
chr29 24945 24961 AAAATTTTGGGGCCCC 100.0 +
JSON Output
After running the tool, a JSON file is generated containing detailed information about the motifs found.
{
"num_blocks": 16393,
"count_of_genomes": 6,
"genome_ids": [
"HG00621#2",
"HG02723#2",
"HG01175#2",
"NA20129#1",
"HG03098#1",
"HG01123#1"
],
"motifs_found": 89,
"motifs": [
{
"hit_id": "pssm_THAP1_1",
"motif_type": "pssm",
"motif_length": 9,
"motif": "CTGCCCGCA",
"gapped_start": 1038,
"gapped_end": 1046,
"ungapped_start": 1038,
"ungapped_end": 1046,
"gapped_sequence": "CTGCCCGCA",
"ungapped_sequence": "CTGCCCGCA",
"conservation": {
"value": 100.0,
"other_genomes": [
{
"genome_id": "CHM13",
"aligned_sequence_gapped": "CTGCCCGCA",
"vector": "111111111",
"conservation": "100.00%",
"gapped_start": 1038,
"gapped_end": 1046
},
{
"genome_id": "GRCh38",
"aligned_sequence_gapped": "CTGCCCGCA",
"vector": "111111111",
"conservation": "100.00%",
"gapped_start": 1038,
"gapped_end": 1046
}
// Additional genome entries...
]
}
}
// Additional motif entries...
]
}
CSV Output
The csv file generated contains the coordinates of the motif found along with its name , type , strand direction and conservation. Each motif_hit is then followed by a number of columns for each aligned sequence in the MAF block where the hit was found. The cells are then filled with an entry, similarity_vector, conservation_score
e.g. 11110110000,54.55%
| motif_hit_info | motif_name | motif_type | motif_strand | score | fake_species_48 | fake_species_45 | fake_species_6 | fake_species_54 | fake_species_65 |
|---|---|---|---|---|---|---|---|---|---|
| chr16:9722-9732,ACCTGTGGTCT | MA0002.2 | pwm | + | 10.025236129760742 | 11110110000,54.55% | 11111111111,100.00% | 11111101111,90.91% | 11011111111,90.91% | 10110111011,72.73% |
| chr46:33765-33775,TAACCACAGAA | MA0002.2 | pwm | + | 11.518963813781738 | 11111111111,100.00% | 11111111011,90.91% | 11111111111,100.00% | 11011011110,72.73% | |
| chr69:51945-51955,TGTTGTGGTTA | MA0002.2 | pwm | + | 10.343249320983887 | 11111111111,100.00% | 01111111111,90.91% | 11111111111,100.00% | 11111011110,81.82% | |
| chr80:61982-61992,CAACCACAGGC | MA0002.2 | pwm | + | 10.74906063079834 | 10111111111,90.91% | 10111111011,81.82% | 11111111101,90.91% |
TMP Directory
To faciliate the parallel processing workflow a temporary directory called tmp is created in the current working directory where the script is run. This directory holds intermediate.tmp files required for the generation of the final output. These files are always equal to the number of workers so there is no possibility of inode resource exhaustion.
Workflow Diagram
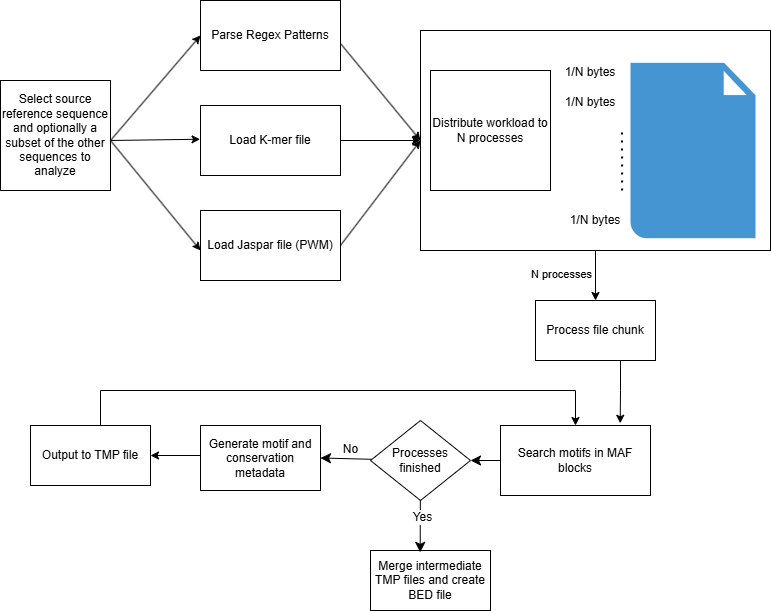
Finding conserved motifs with MAFin
MAFin is a tool that helps identify conserved motifs across multiple genomes by comparing aligned sequences in MAF files. These motifs can be found using one of three methods:
- Regular Expression (regex): A pattern search within the sequence.
- Position Weight Matrix (PWM): A motif search using a predefined PWM.
- K-mer Input: Using specific k-mers to search the sequences.
In the following examples, we will assume the motif being searched for is ATCG.
Conservation Process
Once the motif has been found, MAFin calculates the conservation across aligned sequences. The process is based on comparing the ungapped sequences within the alignment, while still respecting the gaps in the MAF file to maintain the alignment structure. The comparison results in a similarity vector and the following rules are followed to compute it: For each aligned pair of characters:
- If both are gaps, ignore that position.
- If the reference is a gap, record a “–”.
- If the reference is not a gap and matches the compared character, record a “1”.
- If the reference is not a gap and does not match the compared character (including when the compared character is a gap), record a “0”.
The conservation logic also includes calculating a conservation percentage, which tells us how conserved the motif is across genomes, based on the number of matches relative to the length of the motif.
Example 1: Conservation logic
The example covers all edge cases when applying the conservation logic algorithm between a pair of sequences.
Motif: ATCGAC
Reference Genome: A – T C G A – C
Compared Genome: A G T G – A – C
For each aligned pair of characters:
- If both are gaps, ignore that position.
- If the reference is a gap and is compared to a character, we record a “–” in the conservation vector.
- If the reference is not a gap and matches the compared character, record a “1”, for matching.
- If the reference is not a gap and does not match the compared character (including when the compared character is a gap), record a “0”, for a mismatch.
Step-by-Step Comparison:
| Position | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| Ref Base | A | - | T | C | G | A | - | C |
| Cmp Base | A | G | T | G | - | A | - | C |
| Result | Match | Gap | Match | Mismatch | Mismatch | Match | Skip | Match |
| Vector | 1 | - | 1 | 0 | 0 | 1 | 1 |
Similarity Vector:
Positions 1, 3, 6, and 8 yield a “1” (match). Positions 4 and 5 yield a “0” (mismatches), and position 2 is recorded as “–” (gap–nucleotide). The gap–gap at position 7 is skipped. Thus, the resulting similarity vector is:
[1, –, 1, 0, 0, 1, 1]
Conservation Percentage:
Here, the vector length is 7 (position 7 is skipped), and the number of matches (i.e. “1”s) is 4. Therefore, the conservation score is computed as:
Conservation = (4/7) × 100% ≈ 57.14%
Genome Coordinates for Example:
In addition to the similarity vector, MAFin supplies the genomic coordinates of the motif. Consider the example above (using 0-indexed coordinates): after comparing the sequences (where gaps in both sequences are skipped), the ungapped reference motif is: A, T, C, G, A, C (a total of 6 bases). If this motif begins at position 1000 (0-indexed) in the reference genome, then its genomic coordinates are reported as:
Start: 1000, End: 1005
[!IMPORTANT]
The End coordinate on BED output files is non inclusive meaning the coordinates above translate to Start: 1000, End: 1006
Reverse Complement Example
Searcing for a motif on the reverse strand generally involves a search for the reverse complement of a sequence.
Searching reverse strand through Regex Patterns
Reference Sequence: ATCGGCA
Regular Expression: C{2}G ( Two times C followed by G )
Due to the complex nature of Regex patterns reversing and complementing the expression is impossible. Thus we reverse and complement the reference sequence and then search for the pattern there.
Reverse complement match of CCG in sequence: TGCCGAT , results in genomic coordinates 3,5:
| 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|
| T | G | C | C | G | A | T |
Searching reverse strand through K-mers
Reference Sequence: ATCGGCA
K-mer: CCG
Searching for a K-mer on the reverse strand is way simpler. We just reverse complement the k-mer sequence and search for it on the existing strand
Reverse complement K-mer: CGG
Which is found at genomic coordinates 3,5:
| 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|
| A | T | C | G | G | C | A |
Searching reverse strand through PWMs (JASPAR format)
PWM:
| Position | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| A | 0.2 | 0.1 | 0.4 | 0.3 |
| C | 0.3 | 0.5 | 0.1 | 0.2 |
| G | 0.4 | 0.3 | 0.4 | 0.5 |
| T | 0.1 | 0.1 | 0.1 | 0.0 |
To search for a motif on the reverse strand, the reverse complement of the PWM must be calculated. The reverse complement of the above PWM will be represented by reversing the order of the positions and replacing each nucleotide with its complement. Therefore, the reverse PWM becomes:
Reverse PWM:
| Position | 4 | 3 | 2 | 1 |
|---|---|---|---|---|
| A | 0.0 | 0.1 | 0.1 | 0.1 |
| C | 0.5 | 0.4 | 0.3 | 0.4 |
| G | 0.2 | 0.1 | 0.5 | 0.3 |
| T | 0.3 | 0.2 | 0.1 | 0.4 |
And the standard process of PWM search is followed afterwards.
Summary
MAFin computes conservation by comparing aligned sequences while respecting gaps to maintain the MAF file alignment. The similarity vector, which has the same length as the motif, represents matches and mismatches, and the conservation percentage provides an overall measure of how conserved the motif is across genomes. MAFin also outputs genomic coordinates for each motif in the reference and aligned genomes, allowing for easy tracking of motifs across genomes.
License
This project is licensed under the GNU GPL v3.
Contact
For any questions or support, please contact



Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file mafin-0.9.tar.gz.
File metadata
- Download URL: mafin-0.9.tar.gz
- Upload date:
- Size: 29.5 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.2 CPython/3.8.20
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
9b930e8610eaa909a695d2d4bc6cc264fb87a16a5e5fc527563fa879b7dd1607
|
|
| MD5 |
cc502e6a80d073a9e0ba02c994f7408e
|
|
| BLAKE2b-256 |
8a1375b840d921cc2d9010b4ae69e851644715907483fdaf4b80d9dc78611922
|
File details
Details for the file MAFin-0.9-py3-none-any.whl.
File metadata
- Download URL: MAFin-0.9-py3-none-any.whl
- Upload date:
- Size: 29.3 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.2 CPython/3.8.20
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
561307de5fe8a0d6f7585c4a8bedaa835fef58cd01f64b025b475d6902efdfb6
|
|
| MD5 |
e4622e753c9b41024391159a7933d06b
|
|
| BLAKE2b-256 |
e2056be5582f0834109e0e06e1700047c9b39c41da3f55e20248876d0838a2c6
|







