Package & software for analysis of nucleotide redundancy within CDS-based pan-genome analyses

Project description



A pan-genome quality control toolkit for evaluating nucleotide redundancy in pan-genome analyses.
Table of Contents
Motivation
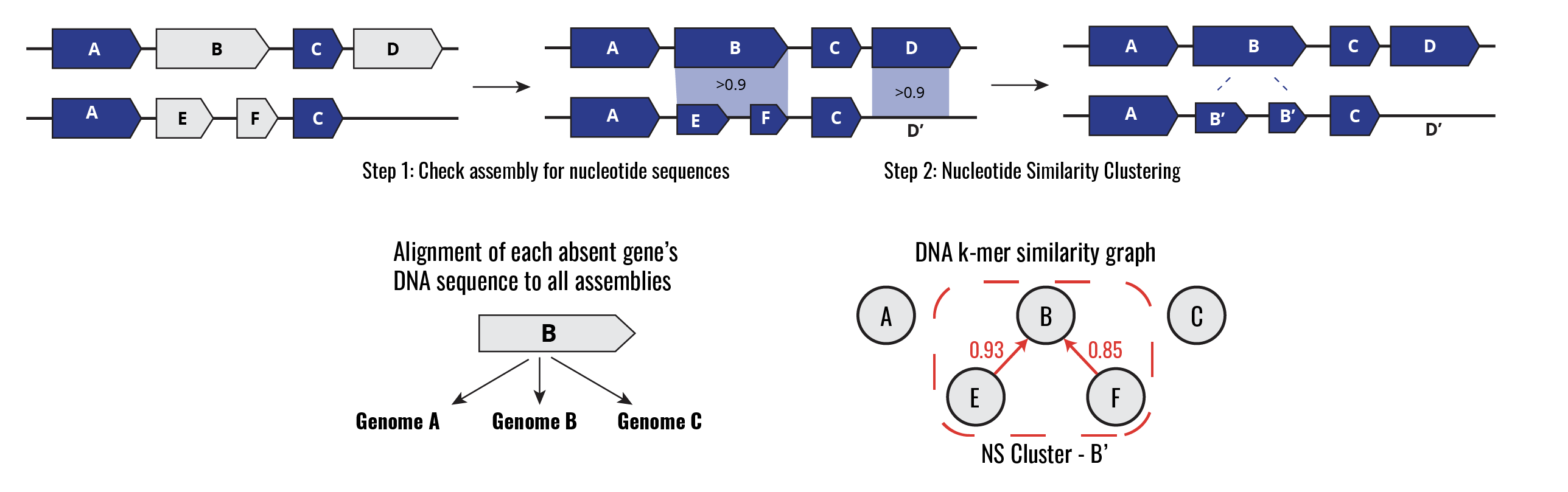
The panqc Nucleotide Redundancy Correction (NRC) pipeline adjusts for redundancy at the DNA level within pan-genome estimates in two steps. In step one, all genes predicted to be absent at the Amino Acid (AA) level are compared to their corresponding assembly at the nucleotide level. In cases where the nucleotide sequence is found with high coverage and sequence identity (Query Coverage & Sequence Identity > 90%), the gene is marked as “present at the DNA level”. Next, all genes are clustered and merged using a k-mer based metric of nucleotide similarity. Cases where two or more genes are divergent at the AA level but highly similar at the nucleotide level will be merged into a single “nucleotide similarity gene cluster”. After applying this method the pan-genome gene presence matrix is readjusted according to these results.
Installation
pip
pip install panqc
Install locally
Currently, panqc can be installed by cloning this repository and installing with pip.
git clone git@github.com:maxgmarin/panqc.git
cd panqc
pip install .
conda
🚧 Check back soon 🚧
Basic usage
panqc nrc -a InputAsmPaths.tsv -r pan_genome_reference.fa -m gene_presence_absence.csv -o NRC_results/
The above command will output an adjusted gene presence absence matrix along with additional statistics to the specified output directory (NRC_results/).
Alternatively, if you would like to use a gene_presence_absence.Rtab file instead of a CSV matrix of gene presence, use add the --is-rtab flag.
panqc nrc -a InputAsmPaths.tsv -r pan_genome_reference.fa -m gene_presence_absence.Rtab --is-rtab -o NRC_results/
Analyzing included test data set
If you wish to run an panqc nrc on an artifical (and abridged) test data set, you can run the following commands:
cd tests/data
# Define path to the 3 needed input files:
# 1) Gene presence absence matrix (As output by Panaroo or Roary)
PG_Matrix_CSV="TestSet1.gene_presence_absence.csv"
# 2) Pan-genome nucleotide reference (As output by Panaroo or Roary)
PG_Ref_FA="TestSet1.pan_genome_reference.fa.gz"
# 3) SampleID + Path for all assemblies used in analysis
Asm_TSV="TestSet1.InputAsmPaths.tsv"
time panqc nrc -a ${Asm_TSV} -r ${PG_Ref_FA} -m ${PG_Matrix_CSV} -o test_results/
NOTE: Make sure that your current working directory (CWD) is tests/data within the repository. The TestSet1.InputAsmPaths.tsv file describes the path to each genome assembly relative to your CWD.
Full usage
panqc has 2 sub-commands:
nrc- Run the full panqc Nucleotide Redundancy Correction pipeline on a pan-genome analyses.utils- Run utlity scripts and sub-pipelines of the full panqc NRC pipeline
panqc nrc
Run the complete panqc Nucleotide Redundancy Correction (NRC) pipeline
$ panqc nrc --help
usage: panqc nrc [-h] -a PathToAsms.tsv -r pan_genome_reference.fasta -m gene_presence_absence.csv -o RESULTS_DIR [-p PREFIX] [-c MIN_QUERY_COV] [-i MIN_SEQ_ID] [-k KMER_SIZE] [-t MIN_KSIM]
optional arguments:
-h, --help show this help message and exit
-a, --asms PathToAsms.tsv
Table with SampleID & Paths to each input assemblies. (TSV)
-r, --pg-ref pan_genome_reference.fasta
Input pan-genome nucleotide reference. Typically output as `pan_genome_reference.fasta` by Panaroo/Roary (FASTA)
-m, --gene_matrix gene_presence_absence.csv
Input pan-genome gene presence/absence matrix. By default is assumed to be a `gene_presence_absence.csv` output by Panaroo/Roary (CSV) If the user provides the --is-rtab flag, the input is assumed to be an .Rtab (TSV)file.
-o, --results_dir RESULTS_DIR
Output directory for analysis results.
-p, --prefix PREFIX
Prefix to append to output files
-c, --min-query-cov MIN_QUERY_COV
Minimum query coverage (ranging from 0 to 1) to classify a gene as present within an assembly (Default: 0.9)
-i, --min-seq-id MIN_SEQ_ID
Minimum sequence identity (ranging from 0 to 1) to classify a gene as present within an assembly (Default: 0.9)
-k, --kmer_size KMER_SIZE
k-mer size (bp) to use for generating profile of unique k-mers for each sequence (Default: 31))
-t, --min-ksim MIN_KSIM
Minimum k-mer similarity (maximum jaccard containment of k-mers between pair of sequences) to cluster sequences into the same "nucleotide similarity cluster" (Default: 0.8))
--is-rtab Flag indicating that the input gene matrix is a tab-delimited .Rtab file
panqc utils
Within utils there are 3 sub-commands that run specific components of the panqc NRC pipeline:
utils asmseqcheck- Perform alignment of all genes classified as absent to their respective assemblies.utils ava- Perform all vs all comparison of k-mer profiles of input sequences.utils nscluster- Perform nucleotide similarity clustering and readjust pan-genome estimates.
$ panqc utils --help
usage: panqc utils [-h] {asmseqcheck,ava,nscluster} ...
positional arguments:
{asmseqcheck,ava,nscluster}
Please select one of the utilility pipelines of the panqc toolkit.
asmseqcheck
ava
nscluster
optional arguments:
-h, --help show this help message and exit
🚧 Check back soon for full usage for each of the utility sub-pipelines of the panqc toolkit 🚧
Contributing and Issues
🚧 Check back soon 🚧
Citing
🚧 Check back soon 🚧
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.







