
SBMLlint
Problem Addressed
Many biological models are based on chemical reactions. For example, glycolysis, arguably the most widely exercised metabolic pathway in biology, begins by transforming the reactants glucosue (Glu) and adenosine triphosphate (ATP) into the products glucose 6-phosphate (GluP) and adenosine diphosphate (ADP), or: Glu + ATP -> GluP + ADP. Examples of biological modeling techniques that rely on reactions include flux balance analysis and kinetics models.
Today's biological models typically consist of tens to hundreds of reactions. Even with this modest level of complexity, it is easy to make mistakes. For example, a mass balance error occurs if the total mass of the reactants differs from the total mass of the products. With the advent of high throughput laboratory techniques, the complexity of models will grow rapidly. As a point of comparison, the complexity of typical software packages has grown from hundereds of lines of code in the 1960s to tens of millions of lines of code for software such as linux and the Apache web server.
Because of this huge growth in the complexity, software engineers developed sophisticated tools to detect errors in codes statically, before any statement is executed. For example, the pylint tool analyzes python source codes to determine if a variable is referenced before a value is assigned to it. The term linter is used for a tool that does static analysis of source codes.
Overview
SBMLLint is a collection of tools for linting reactions. The initial focus is detecting mass balance errors. The tool takes as input a model expressed in either SBML (Systems Biology Markup Language, a standard format for biochemical models) or the Antimony language (a human readable representation of chemical reaction models).
SBMLLint implements two algorithms for detecting mass balance errors. The first, moiety analysis, checks for balance in
the moiety structure of reactions.
For example ATP has the moeities adenosine with three inorganic phosphates.
Moiety analysis requires that modelers follow a naming convention that exposes the moiety structure.
There is no restriction on the choice of moiety names (other than compliance with SBML naming standards).
Moiety names can be exposed in two ways. The first is by using a naming convention.
For example, the modeler could use A to indicate a adenosine moiety and Pi for inorganic phosphate.
Moiety analysis requires that moieties be separated by an underscore (\_).
That is, ATP would be written as A_Pi_Pi_Pi
Similarly, GluP would be written as Glu_Pi. Thus, the above reaction is
written as Glu + A_Pi_Pi_Pi -> Glu_Pi + A_Pi_Pi.
A second way to expose moiety names is through explicit declarations in a configuration file. An example of this is
ATP:
- A, 1
- Pi, 3
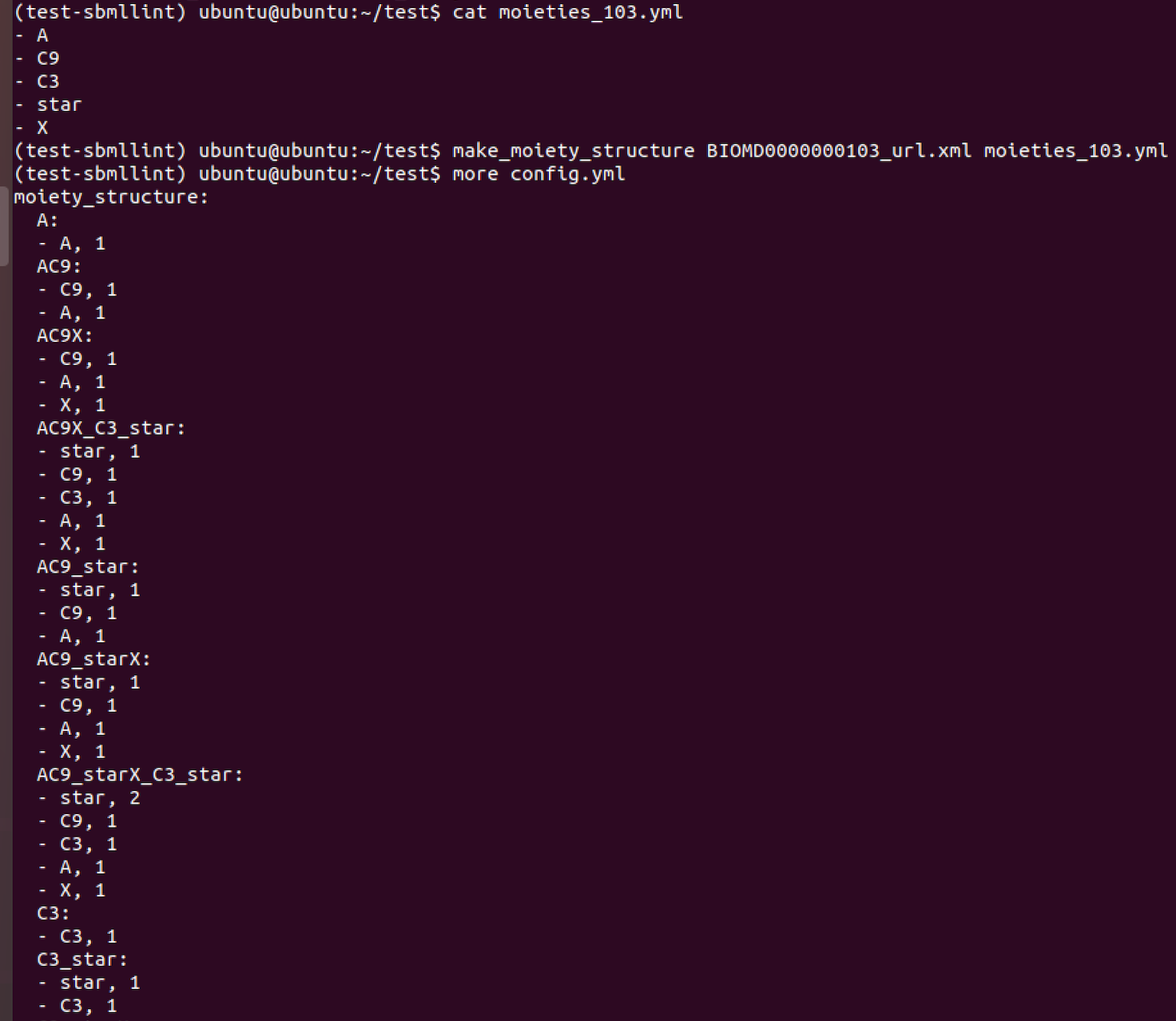
We provide a tool to partially automate the construction of these explicit declarations (make_moiety_structure).
Moiety analysis checks that the count of each moiety in the reactants is the same as the count of each moiety in the products. Although moiety analysis places a burden on the modeler to use the underscore convention, we note that about 20% of the models in the BioModels repository already use names that are close to this structured.
The second algorithm, GAMES (Graphical Analysis with Mass Equality Sets) does not impose any requirements on
the structure of the molecule names.
GAMES checks for stoichiometric inconsistency, which is a weaker condition
than mass balance.
A collection of reactions is stoichiometrically inconsistent if the set of reactions infers that a molecule has a mass of zero. To illustrate this, consider a model consisting of two reactions: A -> B and A -> B + C. The first reaction implies that the mass of A is the same as B. The second reaction implies that the mass of A equals the sum of the masses and B and C. Both statements can be true only if the mass of C is zero, and so the model has a stoichiometric inconsistency.
Using the Tools
SBMLLint provides the following command line tools that are available when SBMLLint is installed.
moiety_analysisuses moiety analysis on an SBML source file to report on mass balance errors.gamesuses games on an SBML source file to report on mass balance errors.make_moiety_structuretakes as input an SBML XML file and a YAML file that lists moiety names to construct the YAML for explicit declarations of moieties.print_reactionstakes as input an SBML XML file and prints the reactions in the model (including their kinetics)lp_analysisdoes linear programming analysis of the model.
The following is an example of using the moiety_analysis and ``GAMES` algorithms to check for mass balance in a Jupyter Notebook.
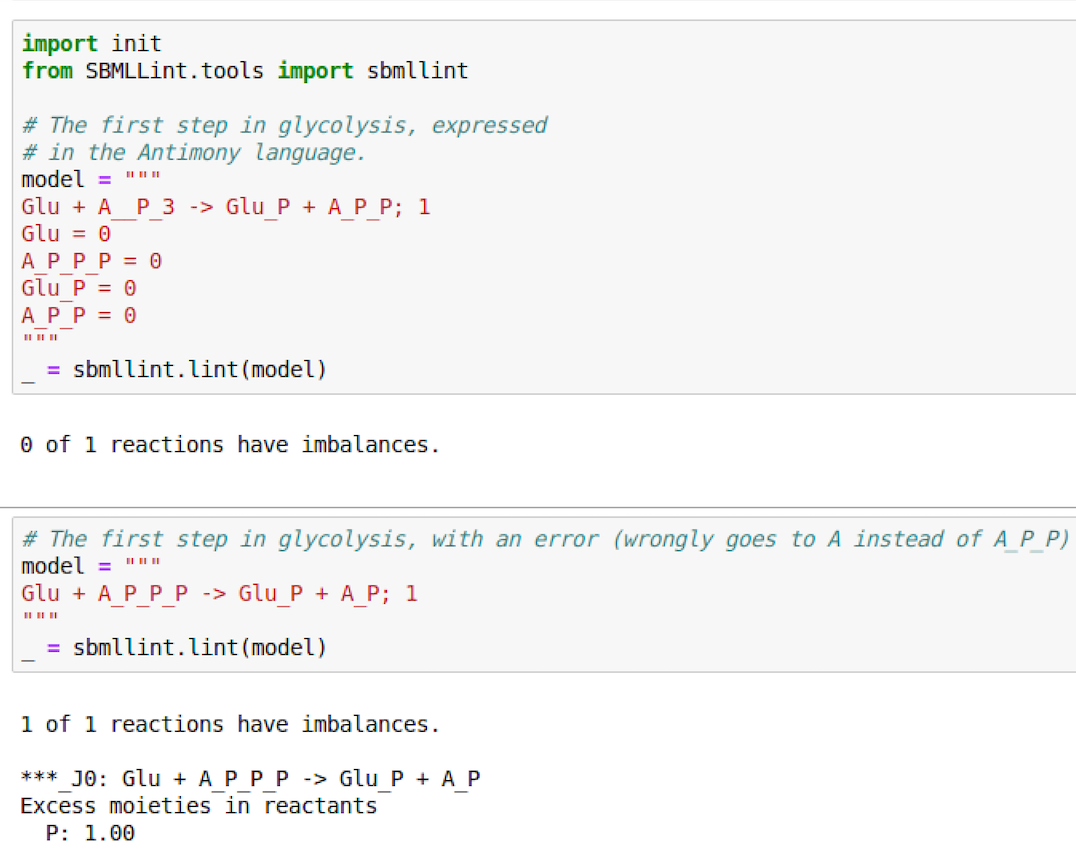

SBMLLint can also be run from the command line, taking as input a model file expressed in SBML or Antimony (if you
install Tellurium).
Below are examples (although the outputs have been truncated).
For moiety analysis:
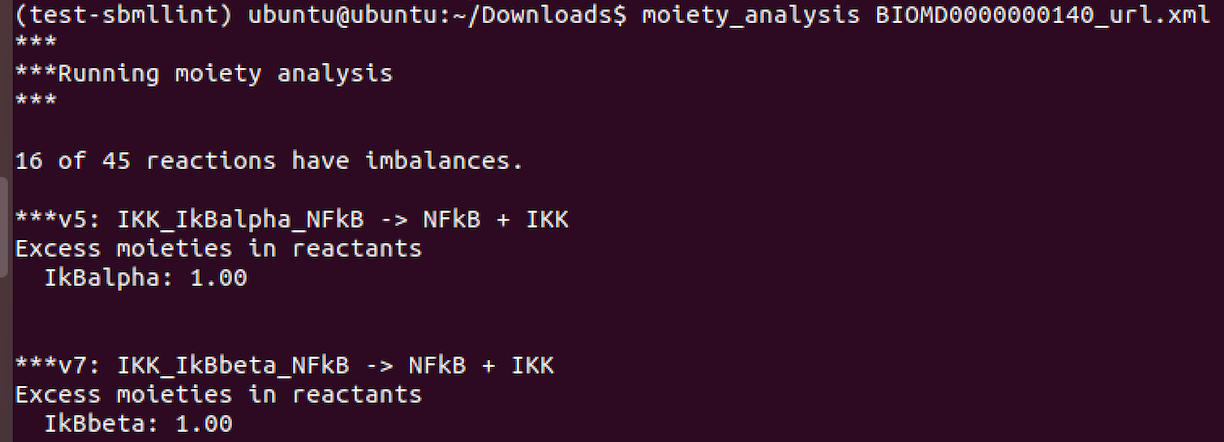
For GAMES:
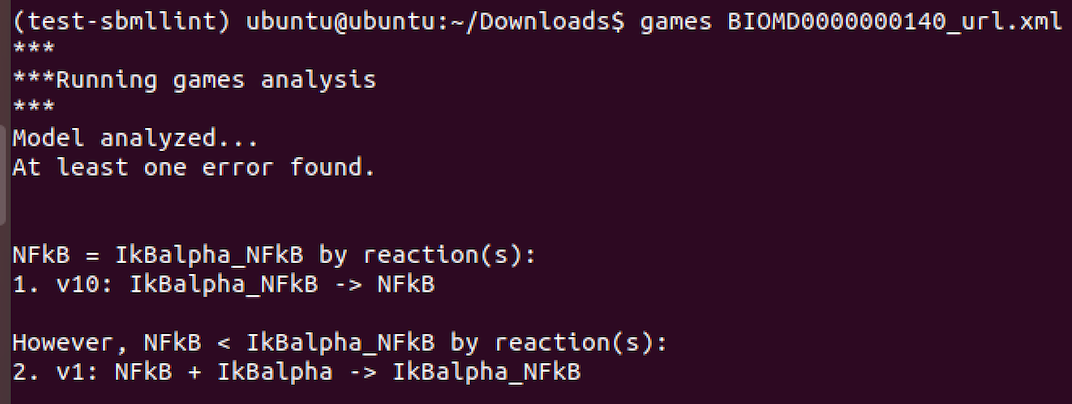
Below we illustrate the use of make_moiety_structure and the format of the YAML input file and the output produced.

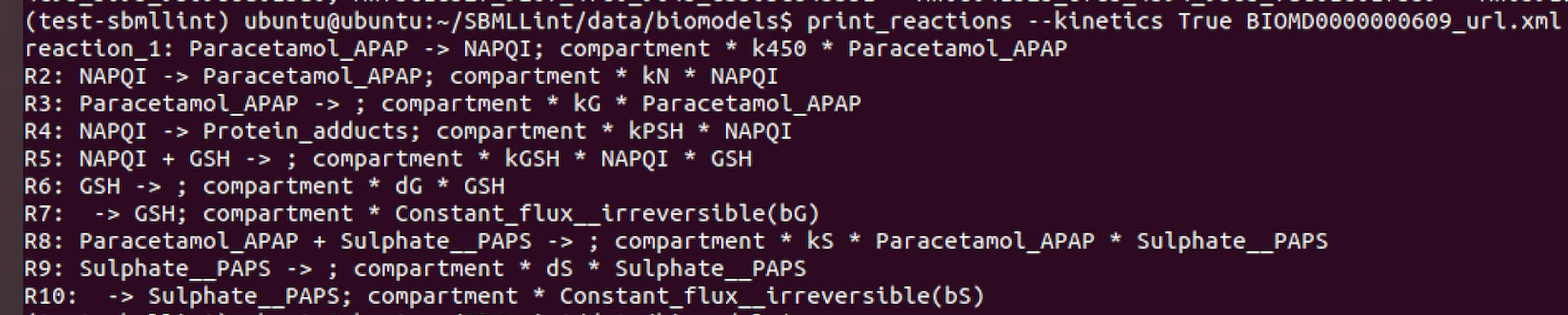
Here is a an example of using print_reactions.
Finally, we provide an example of using lp_analysis.

Installation
SBMLLint is distributed through PyPI. You can install using pip install SBMLLint.
To verify the installation:
- Clone the repository using
git clone https://github.com/ModelEngineering/SBMLLint.git - Set the environment variable PYTHONPATH to the full path to the respository.
nosetests SBMLLint/testson Mac and Linux;nosetests SBMLLint\testson Windows.
Some notes:
-
Depending on your environment, you may see some warning messages, but there should be no errors.
-
The pip install does not include tellurium, and so by default you cannot analyze Antimony files. If you want to analyze Antimony files, you can either install tellurium separately, or
python setup_tellurium.py installin the repository. -
On Windows:
-
You'll need to find where Windows put the installed tools. An easy way to do this is to look at the messages after
pip install SBMLLint. -
Running the SBMLLint tools requires invoking python from the command line using the
pythoncommand. You may need to create a filepython.batthat points to your python installation.
SBMLLint configuration file
SBMLLint can optionally be used with a configuration file. An example of the file can be found in the SBMLLint github folder SBMLLint/.sbmllint_cfg.yml. This is shown below:

The sections of the configuration file are the top level tags that end in a colon (":"). The first two sections provide examples of ignoring molecules and moieties in moiety analysis. The next section indicates whether boundary reactions are considered. By default, boundary reactions are not considered since, by definition, they create or destroy mass. The last section provides for explicit declarations of moiety structures.
The configuration file can be specified in the moiety_analysis and games tools by specifying the ```--config`` option.
Development Notes
The repository contains several shell scripts that add in code development.
install.shinstalls the code in the test-sbmllint virual environment.setup\_env.shsets up the environment variables (use sourcesetup_env.sh).
1 maintainer
Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
File details
Details for the file SBMLLint-1.1.2.tar.gz.
File metadata
- Download URL: SBMLLint-1.1.2.tar.gz
- Upload date:
- Size: 10.3 MB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.4.2 importlib_metadata/4.6.4 pkginfo/1.7.1 requests/2.26.0 requests-toolbelt/0.9.1 tqdm/4.62.2 CPython/3.6.9
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
f700b9f03975a0c3ebb170d7b2242a80101a3af8f585518f60a9ed47ec0b49ac
|
|
| MD5 |
5825fdcbadd1717305b1cf1c15db880c
|
|
| BLAKE2b-256 |
d89c9ec74d59b2d29b63715007f534b93c82bf0600af292275155b29f58103f5
|









