automatically design sgRNA for exon skipping with many base editors

Project description
AltEx-BE: Alternate Exon Skipping by Base Editing





- AltEx-BE: Alternate Exon Skipping by Base Editing
- License
Important Notices
- Japanese version of README is also available! Please visit this file
[!IMPORTANT] WEB version of AltEx-BE is also available! You can use it without installing anything on your computer. Please visit this webpage
Overview
AltEx-BE is a command-line bioinformatics tool that designs sgRNAs (single guide RNAs) to induce targeted exon skipping using Base Editing technology.
Manipulating alternative splicing is key to understanding diseases like cancer and neurodegenerative disorders, but designing the right tools for the job is a major bottleneck. The manual process of identifying targetable exons, designing sgRNAs for specific base editors, and assessing off-target risks is complex, tedious, and slows down critical research.
AltEx-BE is a powerful command-line tool built to automate this entire workflow. It intelligently parses transcript data to find the best exon targets, designs candidates for a multitude of base editors, and evaluates their off-target risk to provide a ranked list of high-confidence sgRNAs.
By transforming a complex, multi-step design process into a single command, AltEx-BE bridges the gap between your scientific question and a successful wet lab experiment, significantly accelerating research into splicing-related diseases and therapies.
Key Features
-
🧬 Automated Target Exon Annotation:
- Automatically parses transcript structures from refFlat files to identify and classify potential targets for exon skipping. This includes Skipped Exons (SE) and exons with Alternative 3'/5' Splice Sites (A3SS/A5SS), eliminating the need for tedious manual searches.
-
⚙️ Universal Base Editor Compatibility:
- Supports virtually any ABE or CBE. You can use built-in presets or define any custom editor by specifying its PAM sequence and editing window, allowing immediate use of the latest editors from new publications.
- AltEx-BE can design sgRNAs for multiple Base-Editors in one run
-
🚀 Streamlined End-to-End Workflow:
- Seamlessly moves from data input to candidate selection. The design command generates sgRNAs, while the visualize command creates comprehensive reports to help you evaluate and rank the best candidates for your experiment.
Workflow Diagram
Here is a simplified diagram illustrating the workflow of AltEx-BE:
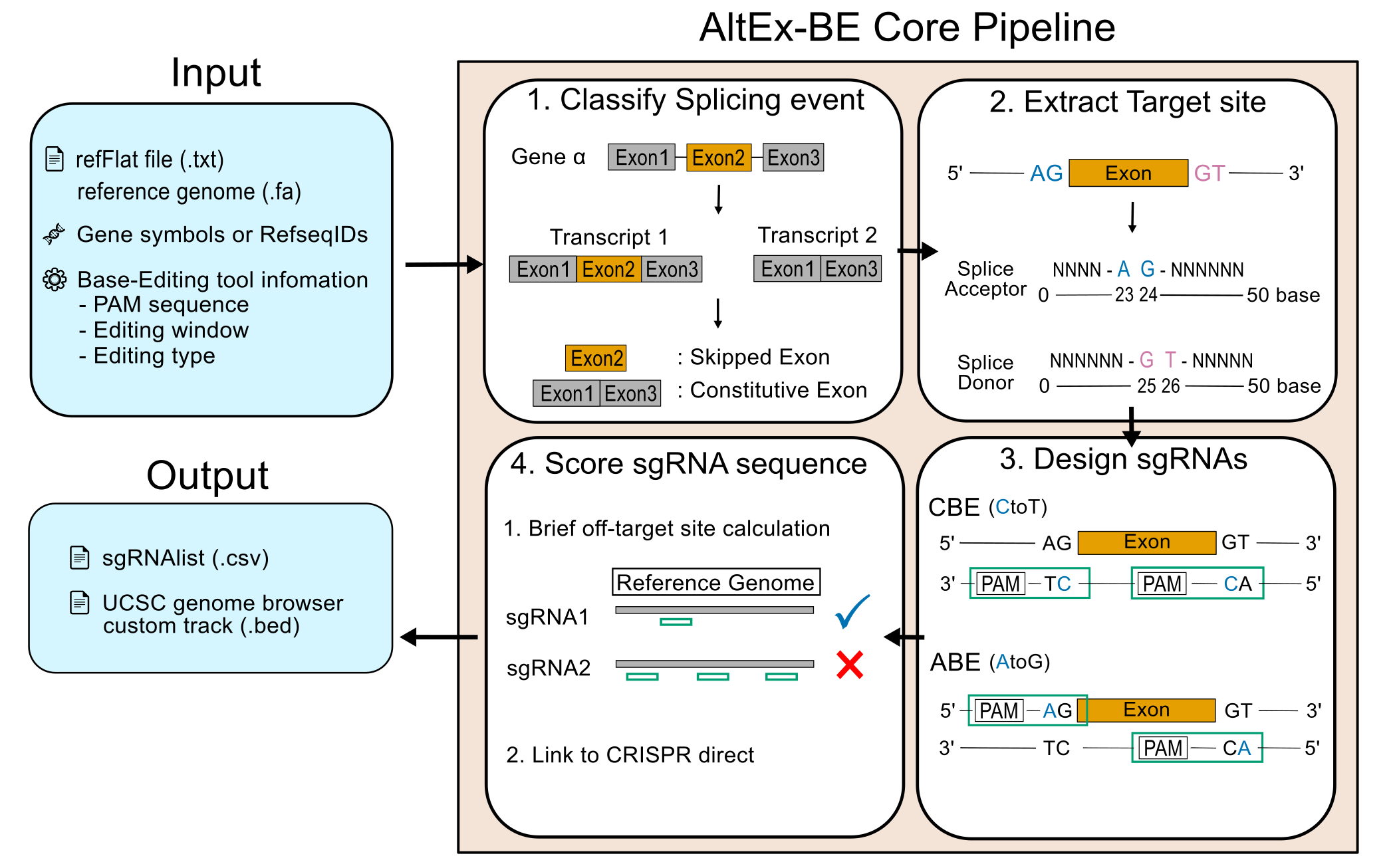
Installation
To get started with AltEx-BE, clone the repository and install the required dependencies.
# 1. install via bioconda
conda install -c conda-forge -c bioconda altex-be
# 2. install via pypi
pip install AltEx-BE
[!CAUTION] AltEx-BE required python 3.10~ 3.12 So when you face to some install errors, please suspect version conflict.
Required dataset
To use AltEx-BE, you should prepare 2 input files in your computer
- refFlat file or gtf file of your interest species
- refflat file contains Refseq infomations: explanation of refFlat format is here
- you can download refflat files from UCSC goldenpath: refflat files of mm39 is here
- also you can use GTF file as a input
- If you use GTF, AltEx-BE automatically converts GTF into refflat format and generate them into output directory
- Fasta files contain all chromosome sequence of your interest species
- you can download Fasta file also from UCSC goldenpath
- please comfirm your .fa files contain all of chromosome. if not, AltEx-BE process will fail
- (optional) CSV or TXT or TSV contain the gene symbols or Refseq IDs
- AltEx-BE is avalilable for many genes. When you want to design sgRNAs for many genes, You can input gene list via
--gene-fileoption. - The input file should only have 1 column with gene symbols or refseq IDs (No need the header row)
- AltEx-BE is avalilable for many genes. When you want to design sgRNAs for many genes, You can input gene list via
[!NOTE] Point of Gene and RefseqID input
- When providing a gene symbol (e.g., MYGENE), AltEx-BE will analyze all known transcripts of that gene to identify alternative splicing events.
- When providing a RefSeq ID (e.g., NM_0012345), AltEx-BE will automatically identify the corresponding gene and analyze all of its transcripts. This ensures a comprehensive analysis even when starting from a single transcript identifier.
Usage
AltEx-BE can be run via a graphical user interface (UI) or directly from the command line.
Graphical User Interface (UI)
For users who prefer a graphical interface, AltEx-BE includes a web-based UI built with Streamlit. It allows you to configure and run the pipeline without using the command line.
To launch the UI, run:
altex-be --ui
The UI helps you:
- 📥 Select input files (FASTA, transcript annotation)
- ⚙️ Configure all options for the analysis
- ▶️ Run the AltEx-BE pipeline locally
- 📝 Monitor execution logs in real time
- 📊 Preview and browse output files
Web Viewer (GitHub Pages)
For a mouse-only workflow, you can use the static web viewer to upload precomputed sgRNA results and browse them in your browser. The viewer is in docs/index.html and works on GitHub Pages because it does not run any computation.
To use it:
- Open
docs/index.htmllocally in a browser, or - Enable GitHub Pages with the
/docsfolder as the source and open the published URL.
Command-Line Interface (CLI)
For command-line usage, AltEx-BE is operated via the altex-be command. Here are a few examples:
1. Using a Preset Editor:
- By default, AltEx-BE design sgRNAs for below 6 Base Editing Tools
[!NOTE] Preset Base Editors:
base_editor_name pam_sequence editing_window_start editing_window_end base_editor_type target_aid_ngg NGG 17 19 cbe be4max_ngg NGG 12 17 cbe abe8e_ngg NGG 12 17 abe target_aid_ng NG 17 19 cbe be4max_ng NG 12 17 cbe abe8e_ng NG 12 17 abe
altex-be \
--refflat-path /path/to/your/refFlat.txt \
--fasta-path /path/to/your/genome.fa \
--output-dir /path/to/output_directory \
--gene-symbols MYGENE \
--assembly-name hg38
2. Input Base Editor Information in the Command Line:
altex-be \
--refflat-path /path/to/your/refFlat.txt \
--fasta-path /path/to/your/genome.fa \
--output-dir /path/to/output_directory \
--gene-symbols MYGENE \
--assembly-name hg38 \
--be-name target-aid \
--be-type cbe \
--be-pam NGG \
--be-start 17 \
--be-end 19
[!CAUTION]
--be-startand--be-endspecify the editing window of your base editor. The location of the editing window is counted from the base next to the PAM (1-indexed).
3. Input a CSV/TSV/TXT File Containing Information about Your Base Editors:
You can provide a file containing the information for one or more base editors. This is useful when you want to design sgRNAs for multiple editors at once.
[!CAUTION] The input file should have the following columns:
base_editor_name,pam_sequence,editing_window_start,editing_window_end,base_editor_type.
altex-be \
--refflat-path /path/to/your/refFlat.txt \
--fasta-path /path/to/your/genome.fa \
--output-dir /path/to/output_directory \
--gene-symbols MYGENE \
--assembly-name hg38 \
--be-files /path/to/your/base_editor_info.csv
List of command line options
| Short Option | Long Option | Argument | Explanation |
|---|---|---|---|
| -h | --help | Show the help message and exit. | |
| -v | --version | Show the version of Altex BE. | |
| --ui | Launch the Streamlit web UI for AltEx-BE. | ||
| -r | --refflat-path | FILE | (Mutually Required -r or -g) Path to the refFlat file. |
| -g | --gtf-path | FILE | (Mutually Required with -r or -g) Path to the GTF file. |
| -f | --fasta-path | FILE | (Required) Path to the FASTA file. |
| -o | --output-dir | DIR | (Required) Directory for the output files. |
| --gene-symbols | SYMBOL [SYMBOL ...] | A space-separated list of gene symbols of interest. | |
| --refseq-ids | ID [ID ...] | A space-separated list of RefSeq IDs of interest. | |
| --gene-file | FILE | Path to a CSV or TXT file contain your interest gene symbols/RefseqIDs | |
| --run-all_genes | store true | when user input this option, AltEx-BE design sgRNAs for all genes | |
| -a | --assembly-name | ASSEMBLY | (Required) The name of the genome assembly to use (e.g., hg38, mm39). |
| -n | --be-name | NAME | The name of the base editor to use. |
| -p | --be-pam | SEQUENCE | The PAM sequence for the base editor. |
| -s | --be-start | INTEGER | The start of the editing window for the base editor (1-indexed from the base next to the PAM). |
| -e | --be-end | INTEGER | The end of the editing window for the base editor (1-indexed from the base next to the PAM). |
| -t | --be-type | TYPE | The type of base editor (ABE or CBE). |
| --be-files | FILE | Path to a CSV or TXT file containing information about one or more base editors. |
Format of AltEx-BE output
altex-be makes 2 output files in Path/To/YourOutput/ directory which you specified in --output-dir command
- Summary sgRNA table (.csv)
- this table contain imformation of sgRNAs designed and ranked by AltEx-BE
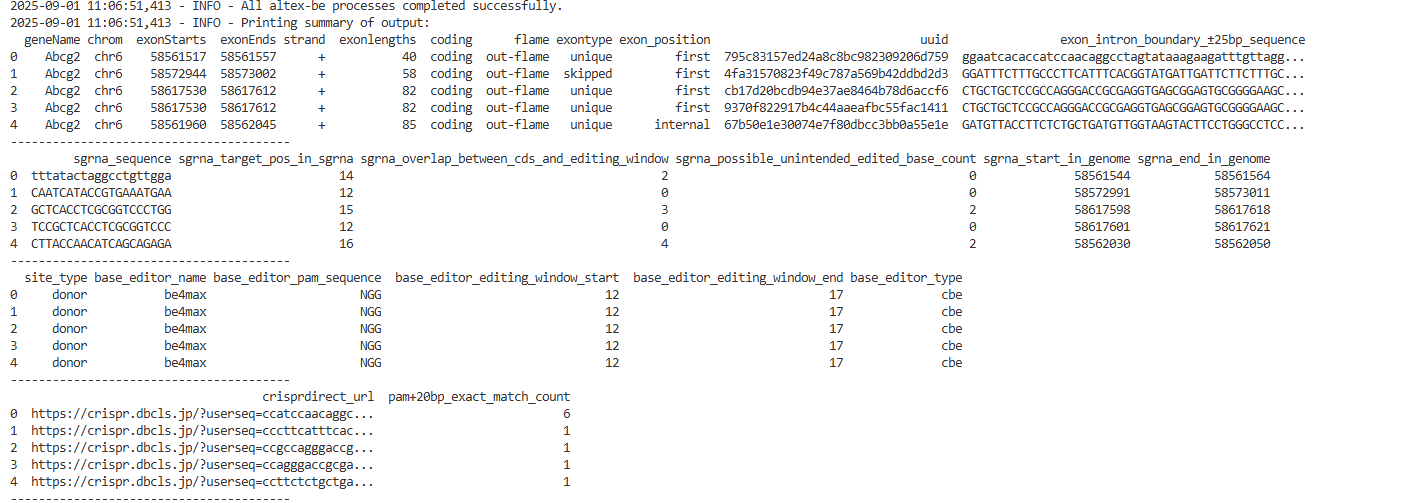 - Meaning of each column is :
- Meaning of each column is :
| column name | meaning | remark |
|---|---|---|
| geneName | gene symbol of target gene | |
| chrom | location of target gene | |
| strand | strand of target gene | |
| exonstart, exonend, exonlength | general information of target exon | |
| coding | whether target gene is protein coding or non coding gene | |
| frame | mod3 of the length of target exon | 0 = in-frame or 1,2 = out-frame |
| exon_position | relative location of target exon in target gene | "first" or "internal" or "last" |
| uuid | the unique id for each sgRNAs | changes in every run |
| exon_intron_boundary+-25bp_sequence | sequence around SA or SD | |
| sgrna_sequence | sgRNA sequence | Thymine is not replaced by Uracil |
| sgrna_target_pos_in_seq | position of target A or C in sgRNA | relative location in sgrna |
| sgrna_overlap_between_cds_and_editing_window | number of overlapping bases with editing window | |
| sgrna_unintended_edited_base_count | number of possible being edited bases (A or C) in cds | |
| sgrna_start/end_in_genome | location of sgrna | |
| site type | target splicing site of sgRNA | acceptor or donor |
| sgrna_strand | strand of sgRNA | |
| base_editor_name/pam_sequence/window_start or end / base editor type | infomation of BE to design sgRNA | |
| crispr_direct_url | link to CRISPR direct | |
| pam+20bp exact match | pam+20bp (23-mer) exact match in all chromosome | |
| pam+12bp exact match | pam+12bp (12-mer) exact match in all chromosome | |
| sgrna_priority | ranking of sgRNA for each target exon | ranked by off-target specificity and GC content |
sgRNA Prioritization
To facilitate the selection of optimal sgRNAs for experimental validation, AltEx-BE ranks sgRNAs for each target splice site based on predicted off-target binding specificity. The tool prioritizes sgRNAs primarily by the number of exact 20-nucleotide matches (PAM + 20bp) across the genome, selecting those with the fewest potential off-target sites. For sgRNAs with equivalent off-target profiles, GC content is considered as a secondary criterion, favoring sgRNAs within the optimal range of 40-60%. In rare cases where multiple sgRNAs remain equivalent, extended off-target matches (PAM + 12bp) and the number of editable bases within the CDS region are used as final tiebreakers.
- BED file for UCSC custom track (.bed)
- this bed file can use as a UCSC custom tracks, you can input that bed file into this webpage
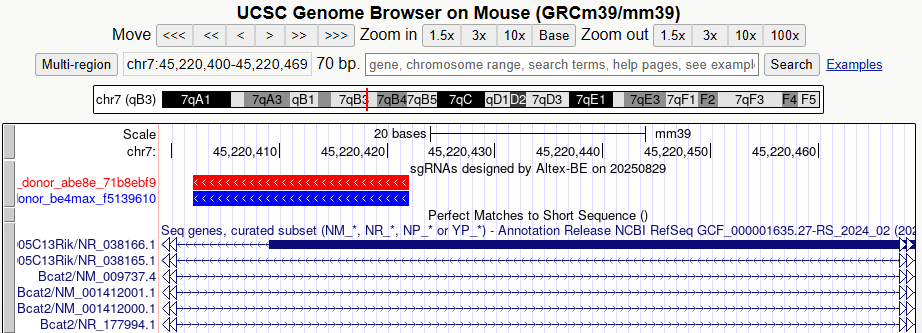 - colored box (red, blue) is sgRNA sequences. red means sgRNAs for abe, blue means sgRNAs for cbe.
- score columns in bed file means offtarget count of 20bp+PAM
- when you assign bed file, you should choose correct assembly name in above website
- colored box (red, blue) is sgRNA sequences. red means sgRNAs for abe, blue means sgRNAs for cbe.
- score columns in bed file means offtarget count of 20bp+PAM
- when you assign bed file, you should choose correct assembly name in above website
License
- Please see LICENSE.md
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file altex_be-1.1.1.tar.gz.
File metadata
- Download URL: altex_be-1.1.1.tar.gz
- Upload date:
- Size: 47.5 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.12.13
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
b0dd1500999298a6670323bce98551f2c7046ff4ebeb71f9426a7f6258574b3b
|
|
| MD5 |
8718cdc450f86eae7e157d486b7843ef
|
|
| BLAKE2b-256 |
e6df513e2f4ec4360e654e8b229910d650391c0a996560f86a445ab90913777f
|
File details
Details for the file altex_be-1.1.1-py3-none-any.whl.
File metadata
- Download URL: altex_be-1.1.1-py3-none-any.whl
- Upload date:
- Size: 51.9 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.2.0 CPython/3.12.13
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
5da18c36660ba5672157abc6398a6d068f6d99068343076ebd3756c784b5761f
|
|
| MD5 |
0f8f87a929823c694637611fc4747999
|
|
| BLAKE2b-256 |
58cd3df7545941ca44596de3a9484281de4f0f1dd2671e8dd263990c8c1388d7
|







