A python package for the distance-based analysis of intra- and inter-protein interactions

Project description
MDInteractions: A python package for the distance-based analysis of intra- and inter-protein interactions.
MDInteractions is a Python package designed to analyze atomic-level intra- and inter-protein interactions from molecular dynamics (MD) simulations. It allows users to specify interaction parameters such as distance thresholds to detect and track contacts between atoms or residues within and between proteins over time.
Installation
To install the package, run:
pip install MDInteractions
Modules
- protein_interactions: The protein_interactions module investigates interactions within a protein group or between two protein groups, identifying residues that are within a user-defined atom-distance threshold at each frame. The final pairs recorded in the CSV file are those that consistently remained within the specified cutoff distance across all of the processed frames.
- mean_distance: The mean_distance module calculates the average distance between all possible pairs of user-defined atoms across one group for intra-protein interactions and across two groups for inter-protein interactions, throughout the user-defined frames of the trajectory.
Example Usage
# Intra-protein interactions: Analyze contacts within a single group
from MDInteractions import protein_interactions
intra_interactions = protein_interactions(
gro_file="md.gro", # Input structure file (.gro)
xtc_file="md.xtc", # Input trajectory file (.xtc)
ndx_file="index.ndx", # GROMACS index file defining atom groups
start_frame=8000, # Frame to start analysis from
end_frame=9000, # Frame to end analysis at
group_ID=10, # Single group ID (intra-protein mode)
group1_atom_name="CB", # One or more atom names as comma-separated string (e.g., 'CB' or 'CB, CA') for first atom set
group2_atom_name="CB", # Same as above, for second atom set
cutoff=7, # Distance cutoff for defining an interaction
residue_specific_atoms=[("GLY", "CA")], # list of residue-specific atom tuples, e.g. '[ ("GLY", "CA"), ("ALA", "CB") ]'
give_res_name=True, # Include residue names in the output
give_atom_name=True # Include atom names in the output
)
intra_interactions.analyze() # Run the interaction analysis
# Inter-protein interactions: Analyze contacts between two distinct groups
from MDInteractions import protein_interactions
inter_interactions = protein_interactions(
gro_file="md.gro",
xtc_file="md.xtc",
ndx_file="index.ndx",
start_frame=8000,
end_frame=9000,
group1_ID=10, # First group ID for inter-protein
group2_ID=11, # Second group ID for inter-protein
group1_atom_name="CB", # One or more atom names as comma-separated string (e.g., 'CB' or 'CB, CA')
group2_atom_name="CB", # Same as above, for group 2
cutoff=15, # Larger cutoff for inter-group interactions
residue_specific_atoms=[("GLY", "CA")], # list of residue-specific atom tuples, e.g. '[ ("GLY", "CA"), ("ALA", "CB") ]'
give_res_name=True,
give_atom_name=True
)
inter_interactions.analyze() # Run the interaction analysis
# Intra-protein mean distance: Average distance of selected atoms within a group
from MDInteractions import mean_distance
intra_meandistance = mean_distance(
gro_file="md.gro",
xtc_file="md.xtc",
ndx_file="index.ndx",
start_frame=8000,
end_frame=9000,
group_ID=10, # Single group ID (intra-protein)
group1_atom_name="CB", # One or more atom names as comma-separated string (e.g., 'CB' or 'CB, CA') for first atom set within group
group2_atom_name="CB", # Same as above, for second atom set within group
residue_specific_atoms=[("GLY", "CA")], # Python-style list of tuples. E.g., '[("GLY", "CA"), ("LYS", "NZ")]'
give_res_name=True, # Include residue names in output
give_atom_name=True, # Include atom names in output
)
intra_meandistance.analyze() # Compute average distances
# Inter-protein mean distance: Average distance between selected atoms across two groups
from MDInteractions import mean_istance
inter_meandistance = mean_distance(
gro_file="md.gro",
xtc_file="md.xtc",
ndx_file="index.ndx",
start_frame=8000,
end_frame=9000,
group1_ID=10, # First group ID for inter-protein mode
group2_ID=11, # Second group ID for inter-protein mode
group1_atom_name="CB", # One or more atom names as comma-separated string (e.g., 'CB' or 'CB, CA')
group2_atom_name="CB", # Same as above, for group 2
residue_specific_atoms=[("GLY", "CA")], # Python-style list of tuples. E.g., '[("GLY", "CA"), ("LYS", "NZ")]'
give_res_name=True,
give_atom_name=True,
)
inter_meandistance.analyze() # Compute average distances
Command Line Interface
# Intra-protein interaction analysis (within one group)
# Input GROMACS .gro structure file
protein_interactions \
--gro_file "md.gro" \
# Input GROMACS .xtc trajectory file
--xtc_file "md.xtc" \
# GROMACS index file defining groups
--ndx_file "index.ndx" \
# Starting frame index for analysis
--start_frame 8000 \
# Ending frame index for analysis
--end_frame 9000 \
# Single group ID (intra-protein)
--group_ID 10 \
# Atom name in group (e.g., beta carbon)
--group1_atom_name CB \
# Atom name for distance comparison (same group)
--group2_atom_name CB \
# Distance cutoff in Ångströms
--cutoff 7 \
# JSON list of residue-specific atom tuples, e.g. '[ ["GLY", "CA", "CB"], ["ALA", "CB"] ]'
--residue_specific_atoms '[["GLY", "CA"]]' \
# Include residue names in output
--give_res_name=True \
# Include atom names in output
--give_atom_name=True
# Inter-protein interaction analysis
# Input GROMACS .gro structure file
protein_interactions \
--gro_file "md.gro" \
# Input GROMACS .xtc trajectory file
--xtc_file "md.xtc" \
# GROMACS index file defining groups
--ndx_file "index.ndx" \
# Starting frame index for analysis
--start_frame 8000 \
# Ending frame index for analysis
--end_frame 9000 \
# First group ID for inter-protein
--group1_ID 10 \
# Second group ID for inter-protein
--group2_ID 11 \
# Atom name for group 1
--group1_atom_name CB \
# Atom name for group 2
--group2_atom_name CB \
# Larger cutoff for inter-group distances
--cutoff 15 \
# JSON list of residue-specific atom tuples, e.g. '[ ["GLY", "CA"], ["ALA", "CB"] ]'
--residue_specific_atoms '[["GLY", "CA"]]' \
# Include residue names in output
--give_res_name=True \
# Include atom names in output
--give_atom_name=True
# Mean distance calculation for intra-protein
# Input GROMACS .gro structure file
mean_distance \
--gro_file "md.gro" \
# Input GROMACS .xtc trajectory file
--xtc_file "md.xtc" \
# GROMACS index file defining groups
--ndx_file "index.ndx" \
# Starting frame index for analysis
--start_frame 8000 \
# Ending frame index for analysis
--end_frame 9000 \
# Single group ID (intra-protein)
--group_ID 10 \
# Atom name in group (e.g., beta carbon)
--group1_atom_name CB \
# Atom name for distance comparison (same group)
--group2_atom_name CB \
# JSON list of residue-specific atom tuples, e.g. '[ ["GLY", "CA"] ]'
--residue_specific_atoms '[["GLY", "CA"]]' \
# Include residue names in output
--give_res_name=True \
# Include atom names in output
--give_atom_name=True
# Mean distance calculation for inter-protein
# Input GROMACS .gro structure file
mean_distance \
--gro_file "md.gro" \
# Input GROMACS .xtc trajectory file
--xtc_file "md.xtc" \
# GROMACS index file defining groups
--ndx_file "index.ndx" \
# Starting frame index for analysis
--start_frame 8000 \
# Ending frame index for analysis
--end_frame 9000 \
# First group ID for inter-protein
--group1_ID 10 \
# Second group ID for inter-protein
--group2_ID 11 \
# Atom name in group (e.g., beta carbon)
--group1_atom_name CB \
# Atom name for distance comparison (same group)
--group2_atom_name CB \
# JSON list of residue-specific atom tuples, e.g. '[ ["GLY", "CA"] ]'
--residue_specific_atoms '[["GLY", "CA"]]' \
# Include residue names in output
--give_res_name=True \
# Include atom names in output
--give_atom_name=True
Parameter Notes
group_IDis used only for intra-protein interactions analysis (single group).group1_IDandgroup2_IDare used only for inter-protein interactions analysis (two groups).group1_atom_nameandgroup2_atom_nameare always required:- For intra-protein interactions, they refer to the atom name or comma-separated atom names of interacting pairs within the same group.
- For inter-protein interactions, they refer to the atom name or comma-separated atom names of interacting pairs between different groups.
- You can specify the same atom name for both groups (e.g.,
"CB") or different atom names (e.g.,"CA"vs"CB") depending on your analysis goals. - The
residue_specific_atomsparameter should be provided as a Python list of tuples (e.g.,[("GLY", "CA"), ("ALA", "CB")]) when using the Python API, and as a JSON-formatted string (e.g.,'[["GLY", "CA"], ["ALA", "CB"]]') when using the command-line interface. - By default, the output file is saved in the same directory as the gro_file, unless an alternative location or file path is specified via the --output_file command-line flag or the output_file="" keyword argument.
Visualising
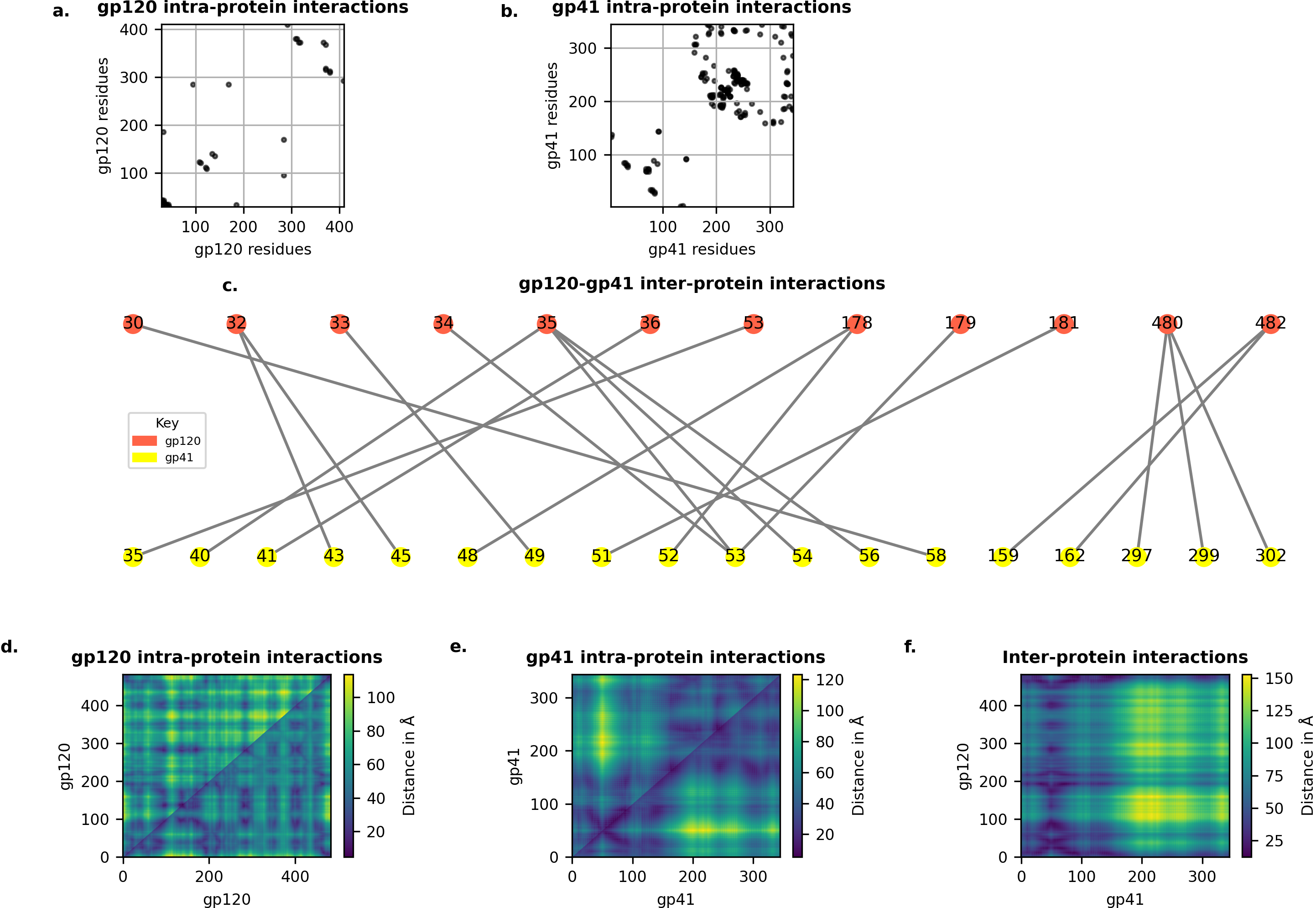
License
This project is licensed under the MIT License.
Citation
If you use MDInteractions in your research, please cite it as:
Fowdar, A and Martin, DP. MDInteractions: A python package for the distance-based analysis of intra- and inter-protein interactions. GitHub, 2025. https://github.com/afowdar/MDInteractions
Or use the following BibTeX entry:
@misc{mdinteractions2025,
author = {Anjani Fowdar and Darren P Martin},
title = {MDInteractions: A python package for the distance-based analysis of intra- and inter-protein interactions},
year = {2025},
howpublished = {\url{https://github.com/afowdar/MDInteractions}},
}
Getting help
For questions, ideas, or feedback, join the conversation in the Discussions section! Feel free to reach out: anjanifowdar@gmail.com
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file MDInteractions-1.1.0.tar.gz.
File metadata
- Download URL: MDInteractions-1.1.0.tar.gz
- Upload date:
- Size: 14.6 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.10.5
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
4801f529fbd213e7116ace49a611a96f4be1f98378fbbdcfe990a155ae237d62
|
|
| MD5 |
68bb997d384a44ded31eb7da0db5682d
|
|
| BLAKE2b-256 |
4c3418307664d52c94a9cf56ff33256a1a45a284d4aa462b53bd8732f8e70fd4
|
File details
Details for the file MDInteractions-1.1.0-py3-none-any.whl.
File metadata
- Download URL: MDInteractions-1.1.0-py3-none-any.whl
- Upload date:
- Size: 19.5 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/6.1.0 CPython/3.10.5
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
8b5648ffd10238b5145b977967cb38dcb0177e97c86677d8bc80dd0901de99c2
|
|
| MD5 |
f498a30e25fb96766027adfd08e7149b
|
|
| BLAKE2b-256 |
58faa125e74f1892d96244f4dc8999e5b62b0c2f9445f18bc32ca3e3c6daddd6
|







