Tool for drawing complex planar molecular systems of arbitrary composition and molecule placement


Project description
MolPainter
Table of Contents
- Introducing MolPainter and MolSolvator
- Installation
- Dependencies
- Test Dependencies
- Documentation
- Tutorial
- What's new in 1.1?
Introducing MolPainter and MolSolvator
Tools for building and solvating complex, planar molecular systems of arbitrary molecular composition and placement via painting.
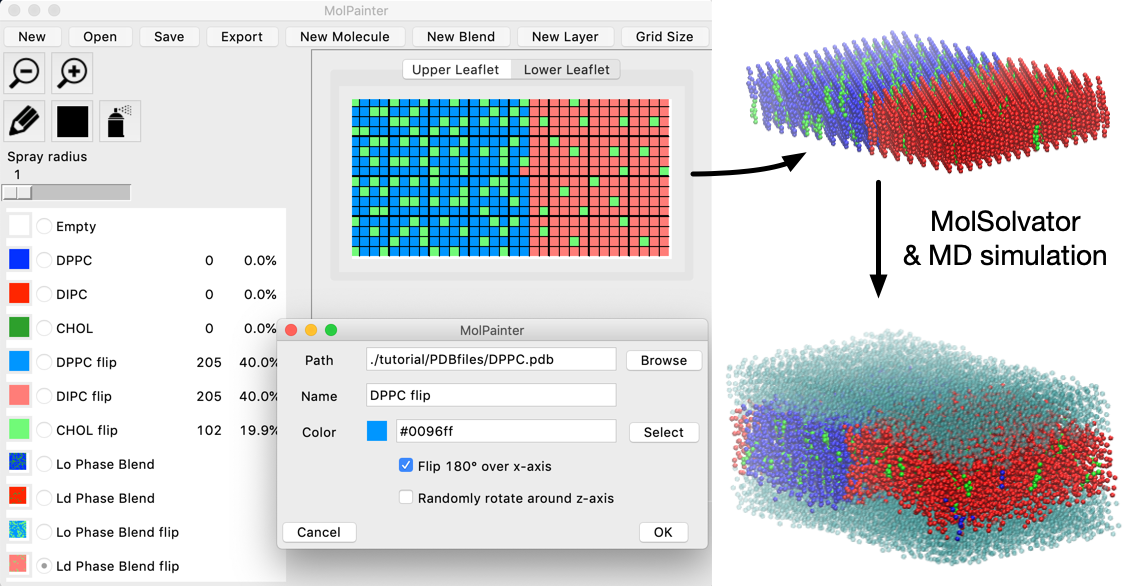
MolPainter is a novel graphical tool that enables users to specifically define the location of molecules in multi-layered, planar molecular systems. MolPainter achieves this by treating each plane of a hypothetical molecular system, defined by a z-axial position, as a two dimensional grids which serve as canvases. By associating molecular structures (in PDB format) to colors, these canvases can be painted to precisely define molecular environments.
MolSolvator, the sister program of MolPainter, is a command line tool that can rapidly solvate such planar systems within the context of the lattices of the "solute" systems produced by MolPainter.
Installation
Install through pip:
pip install MolPainter
Run from the command line:
molpainter
molsolvator -i input.toml
Dependencies
MolPainter's GUI makes use of tkinter in order to provide a native interface on Linux, Mac, and Windows.
The additional package dependencies are numpy, scipy, toml, and MDAnalysis.
Test Dependencies
Tests can be run with pytest from the root of the source tree:
python3 -m pytest
Documentation
Descriptions of the objects and functions of MolPainter and MolSolvator are available here
Tutorial
A tutorial demonstrating the major functions of MolPainter and MolSolvator on a complex mixture lipid bilayer is available here
What's new in 1.1?
Solute systems can now be added during a MolPainter session! These are systems in PDB format to which you want to paint using MolPainter while avoiding clashes.
Let's say for example you wish to use MolPainter to paint lipids around one or multiple proteins. In 1.0 you would have to do this by guessing where the protein will be located and paint the system to try to avoid clasing with the protein. Not very convenient...
Now you can Insert a Solute into MolPainter canvases. A solute will "obstruct" cells in the layers of MolPainter, making them unavailable for painting, based on the real coordinates of the Solute system described in the input PDB file. Just how far these obstructed cells extend from the coordinates of the Solute can be tweaked by adjusting the Buffer space. This is the same kind of "buffer" used in MolSolvator to avoid clashes when solvating.
When the system is ultimately exported, the solute will be written first, followed by the molecules that have been painted using MolPainter.
The new Insert Solutes button has the following fields
- Path path to a PDB file of the solute you want to add. There can only be one solute in the system at a time!
- Buffer space (Å) the length of the buffer added to obstruct more cells neighboring coordinates of the solute.
- Center solute at z (Å) centers the (x,y,z) coordinates of the solute to the center of the MolPainter grid at a particular plane in the z-dimension.
- Expand grid to fit solute expands the MolPainter grid to fit the (x,y,z) coordinates of the solute system. If the solute system has negative coordinates, these coordinates will still lay outside of the MolPainter canvases!
Let us promote your work using MolPainter!
- Please reach out to us and let us know about the cool work you've done!
- We would like to promote interesting systems constructed using MolPainter.
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file MolPainter-1.1.6.tar.gz.
File metadata
- Download URL: MolPainter-1.1.6.tar.gz
- Upload date:
- Size: 38.3 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.2 CPython/3.9.16
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
6de37801b2b15458fa31f0c11ca9a8dda0bb2fea948a70408188302e57fd9ab9
|
|
| MD5 |
8a51a730588d1a465adbaed3182591db
|
|
| BLAKE2b-256 |
9b9432b1252467bed81cfa0a00f3faaa26a44e210e879bc4cb0334f1b7a493f4
|
File details
Details for the file MolPainter-1.1.6-py3-none-any.whl.
File metadata
- Download URL: MolPainter-1.1.6-py3-none-any.whl
- Upload date:
- Size: 52.2 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/4.0.2 CPython/3.9.16
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
af985f8e6a68d55f03e769c8661b855c103f716f2427d143ec0bc26246bca479
|
|
| MD5 |
4a71e83d7ee849c4badfce478ad1bdf0
|
|
| BLAKE2b-256 |
41d6455be9d145781981bb35c56a3686af5a6072639af266a5a4000fc3caed20
|







