ProtParCon - A framework for framework for processing molecular data and identifying parallel and convergent amino acid replacements.

Project description
ProtParCon
ProtParCon is an application framework for manipulating molecular data and identifying parallel and convergent amino acid replacements at the molecular level. Although ProtParCon was not designed for implementing new methods or algorithms for molecular data manipulation, ProtParCon integrates several widely used programs for multiple sequence alignment (MSA), ancestral states reconstruction (ASR), protein sequence simulation, Maximum-Likelihood tree inference (ML Tree) and molecular convergence identification. Therefore, it can be used as a general tool to do MSA, ASR, and simulation under a common interface by using various pre-existed programs under hood.
Work-Flow of ProtParCon
ProtParCon processes a set of orthologous protein sequences with known phylogenetic relationships in six stages:
MSA
ASR
IDENTIFY
SIM
IDENTIFY,
TEST
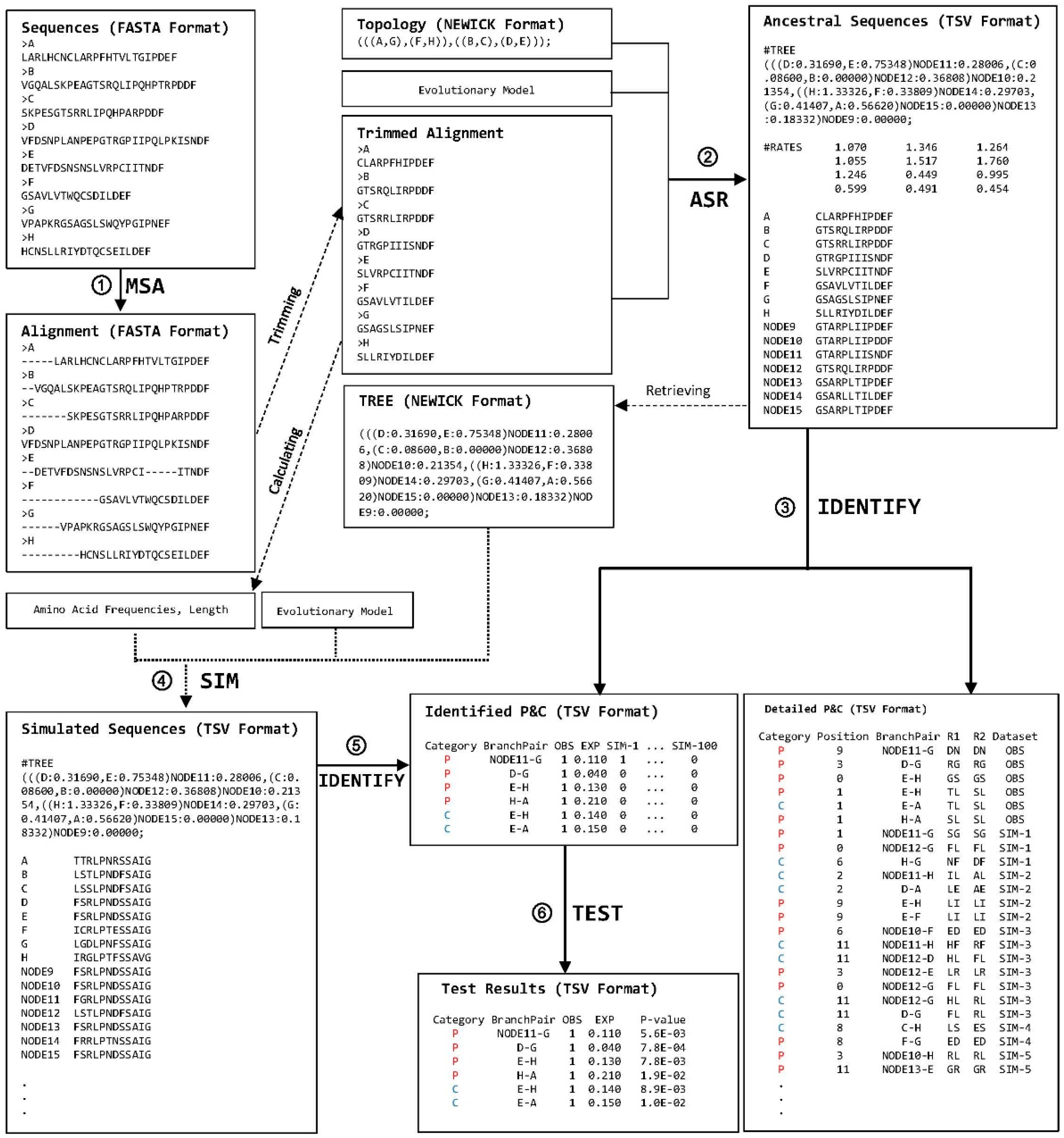
Overview of the ProtParCon analytical scheme
During the multiple sequence alignment (MSA) stage, protein sequences are aligned while gaps and ambiguous character states are trimmed.
In the ancestral state reconstruction (ASR) stage, ancestral character states at each site are inferred for each internal node in the reconstructed tree.
Observed parallel and convergent amino acid replacements for pairs of branches are identified in the IDENTIFY stage. Parallel replacements are denoted by P (red) and convergent replacements by C (blue).
Simulations are conducted in the SIM (simulation) stage. Simulated sequences are evolved according to the following parameters:
an evolutionary model (a replacement rate matrix)
the branching pattern and branch lengths of the tree estimated in the ASR stage
amino acid frequencies and sequence length estimated from the trimmed alignment.
Expected parallel and convergent replacements are identified after the SIM stage or they are directly calculated if no simulation is conducted.
The differences between numbers of observed and expected parallel and convergent replacements for branch pairs of interest are tested during the TEST stage. For better readability, only part of simulated sequences and detailed P&C data are shown. TSV (Tab Separated Values) format data are reformatted. Notation of branch pair, A-B, means a branch pair involving two branches that are leading to A and B, respectively. R1 and R2 represent two amino acid replacement events along two branches. The standard one-letter abbreviations for amino acids is used for the replacements.
Walk-through of an example
In order to show you what ProtParCon brings to the table, we’ll walk you through an example using the simplest way to identify parallel and convergent amino acid replacements at the protein sequence level.
Here is the code for ProtParCon identifying parallel and convergent amino acid replacements within an orthologous protein:
from ProtParCon import imc sequence = 'path/to/the/orthologous/protein/sequence' tree = 'path/to/the/phylogenetic/tree' muscle = 'path/to/the/executable/of/muscle/alignment/program' codeml = 'path/to/the/executable/of/codeml/program' evolver = 'path/to/the/executable/of/evolver/program' imc(sequence, tree, aligner=muscle, ancestor=codeml, simulator=evolver)
Put the above code in a text file, name it something like imc_analyze.py and run the script using Python in a terminal:
$ python imc_analyze.py
Wait for this to finish you will have six files in your work directory: msa.fa, trimmed.msa.fa, ancestors.tsv, simulations.tsv, imc.counts.tsv, and imc.details.tsv. From their names, you may already know what contents in these files. The imc.counts.tsv contains the number of parallel and convergent amino acid replacements that have been identified among all comparable branches, and it looks like this (reformatted here for better readability):
Category BranchPair OBS SIM-1 SIM-2 SIM-3 SIM-4 SIM-5
P A-B 0 0 1 1 0 1
P A-NODE10 0 0 0 0 0 0
P A-NODE11 3 2 1 0 3 2
P A-NODE13 0 2 0 1 0 2
P A-E 0 0 0 0 0 0
C A-B 0 2 1 2 2 0
C A-NODE10 0 0 0 0 0 0
C A-NODE11 0 0 0 1 1 0
C A-NODE13 0 0 1 2 0 1
The imc.details.tsv contains the details of parallel and convergent amino acid replacements that have been identified, e.g. replacement occurred between which branch pairs, on which position of the protein sequence, what kind of replacement, and so on.
What just happened?
When you run the script, imc() look for a sequence file and pass it to a multiple sequence alignment program (MUSCLE program was used in this example), after done with the sequence alignment, imc() look for a phylogenetic tree file and pass it along with the alignment (already removed all gaps and ambiguous characters) to a ancestral sequence reconstruction program (CODEML program inside PAML package is used) to infer the ancestral states. Since a simulation program (EVOLVER program inside PAML package) is specified via argument simulator, ProtParCon will automatically prepare all files needed by evolver and then use evolver to conduct sequence simulation. Once imc() all these works are done, it will start to identify parallel and convergent amino acid replacements along the protein sequence and finally save the results to text files.
Here you notice that one of the main advantages about ProtParCon: sequence alignment, ancestral states reconstruction, and sequence simulation are automatically done without users calling each program step by step. This means ProtParCon already have a pipeline that chained all these processes together, users are only required to tell ProtParCon how they want the sequence to be handled and what results they want to get. Another advantage of using ProtParCon is that it provides a common interface for all supported programs, users no longer need to learn how to use the program and handle the results of these programs.
While ProtParCon enables users to do very fast parallel and convergent amino acid replacement identifications (by use a single sequence file and a tree file) , ProtParCon also gives users full control of the identification process through explicitly manage the workflow step by step. Users are able to do things like choosing preferred sequence alignment program to get high quality sequence alignment, passing more parameters to ancestral states reconstruction program to get accurate ancestral states, and getting full control of sequence simulation process by explicitly using the simulation module with additional options.
What else?
You’ve seen how to run fast parallel and convergent amino acid replacement identifications using general function imc() in ProtParCon package, but this is just the surface. ProtParCon provides a lot of powerful features for manipulating molecular data and makes parallelism and convergence identification even phylogenetic analysis much easier and more efficient, such as:
Built-in support for a lot of sequence alignment programs for multiple sequence alignment (MSA) using simple function.
Built-in support for a lot of phylogenetic tree inference programs for inferring best maximum likelihood tree using simple function.
Built-in support for a lot of ancestral states reconstruction programs for ancestral states reconstruction (ASR) using simple function.
Built-in support for a lot of sequence simulation programs for simulating sequences under various evolutionary scenarios using simple function.
Built-in support for identifying parallel and convergent amino acid replacements using raw orthologous sequence, multiple sequence alignment, reconstructed ancestral sequences, or even simulated sequences.
What’s next?
The next steps for you to do: install ProtParCon, follow through the pre-made examples to learn how to unleash the full power of ProtParCon, use ProtParCon in your routine work to ease the process of molecular data manipulation and molecular parallelism and convergence identification, and finally extend ProtParCon to make it support more and more programs if you are interested in ProtParCon. Thanks for you interest!
See the full description and documentation of ProtParCon for more details!


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file ProtParCon-3.1.tar.gz.
File metadata
- Download URL: ProtParCon-3.1.tar.gz
- Upload date:
- Size: 101.3 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/1.13.0 pkginfo/1.4.2 requests/2.21.0 setuptools/40.8.0 requests-toolbelt/0.9.1 tqdm/4.28.1 CPython/3.6.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
da6f8bdf8afbbf1db3861372af0b090961024282a92d4de020483d97bfbc1f79
|
|
| MD5 |
9fe0ea03844f7df4e8c404bd19e2b3ce
|
|
| BLAKE2b-256 |
30b2c3040e39cce4511cf30ceccb4e25ac5fa30cb7d8449c673f8a9695ea4040
|
File details
Details for the file ProtParCon-3.1-py3-none-any.whl.
File metadata
- Download URL: ProtParCon-3.1-py3-none-any.whl
- Upload date:
- Size: 116.6 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/1.13.0 pkginfo/1.4.2 requests/2.21.0 setuptools/40.8.0 requests-toolbelt/0.9.1 tqdm/4.28.1 CPython/3.6.7
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
471389f5a5dbc06d893c0c946789e36b96027871c1a596e5c6ceb91acab1d8a2
|
|
| MD5 |
78d7cfc523e28075a81ce8caeb521b7f
|
|
| BLAKE2b-256 |
d3b9f07c927a5398e2170558d065ec173204cc119ef3a7709b584c15dddbb7da
|







