Integrated pipeline for ML phylogenetic inference from ABI trace and FASTA data

Project description
AB12PHYLO



AB12PHYLO is an integrated, easy-to-use pipeline for Maximum Likelihood (ML) phylogenetic tree inference from ABI traces and FASTA data.
At its core, AB12PHYLO runs parallelized instances of RAxML-NG (Kozlov et al. 2019) or IQ-Tree (Nguyen et al. 2015) as well as a BLAST search in a reference database.
It enables visual, effortless sample identification based on phylogenetic position and sequence similarity, as well as population subset selection aided by metrics like Tajima's D for estimations of ongoing evolution, or definition of haplotypes.
Documentation
There are two versions of AB12PHYLO, both started from a terminal: ab12phylo as a graphical user interface intended to be more user-friendly and intuitive, and in some details more powerful than ab12phylo-cmd. This version, on the other hand, is a commandline-only tool for maximum reproducibility and automation of a linear pipeline.
While ab12phylo comes with its own on-screen help, and a very brief example for ab12phylo-cmd is provided below, detailed installation and usage instructions can be found in the github wiki. Especially for the commandline ab12phylo-cmd, also check the in-line help via ab12phylo-cmd -h.
For more individual support or feature requests, please write an email to ab12phylo@gmail.com.
Installation
AB12PHYLO can be installed using conda or pip:
conda install -c lkndl -c conda-forge -c bioconda ab12phylo
or
pip install ab12phylo
| :memo: | WINDOWS USERS |
|---|
Windows users must use Anaconda, and run ab12phylo-init before starting the graphical ab12phylo!
When AB12PHYLO is first run, it will check the system for three important non-python tools: RAxML-NG, IQ-Tree 2 and BLAST+. If they are not installed or outdated, AB12PHYLO can download the latest static binaries from GitHub or the NCBI respectively. Check the wiki for more details, troubleshooting, installing from source or updating the package.
As implied above, start the graphical version via ab12phylo from the terminal, and invoke the commandline version via ab12phylo-cmd.
Quick start and functionality
ABI trace files are the main input for AB12PHYLO. Additionally, wellsplate tables can be used to translate back to original sample IDs, provided the mapping is identical for all sequenced genes. Reference data may be included in FASTA format, and the graphical AB12PHYLO accepts FASTA sequences as the main input format as well.
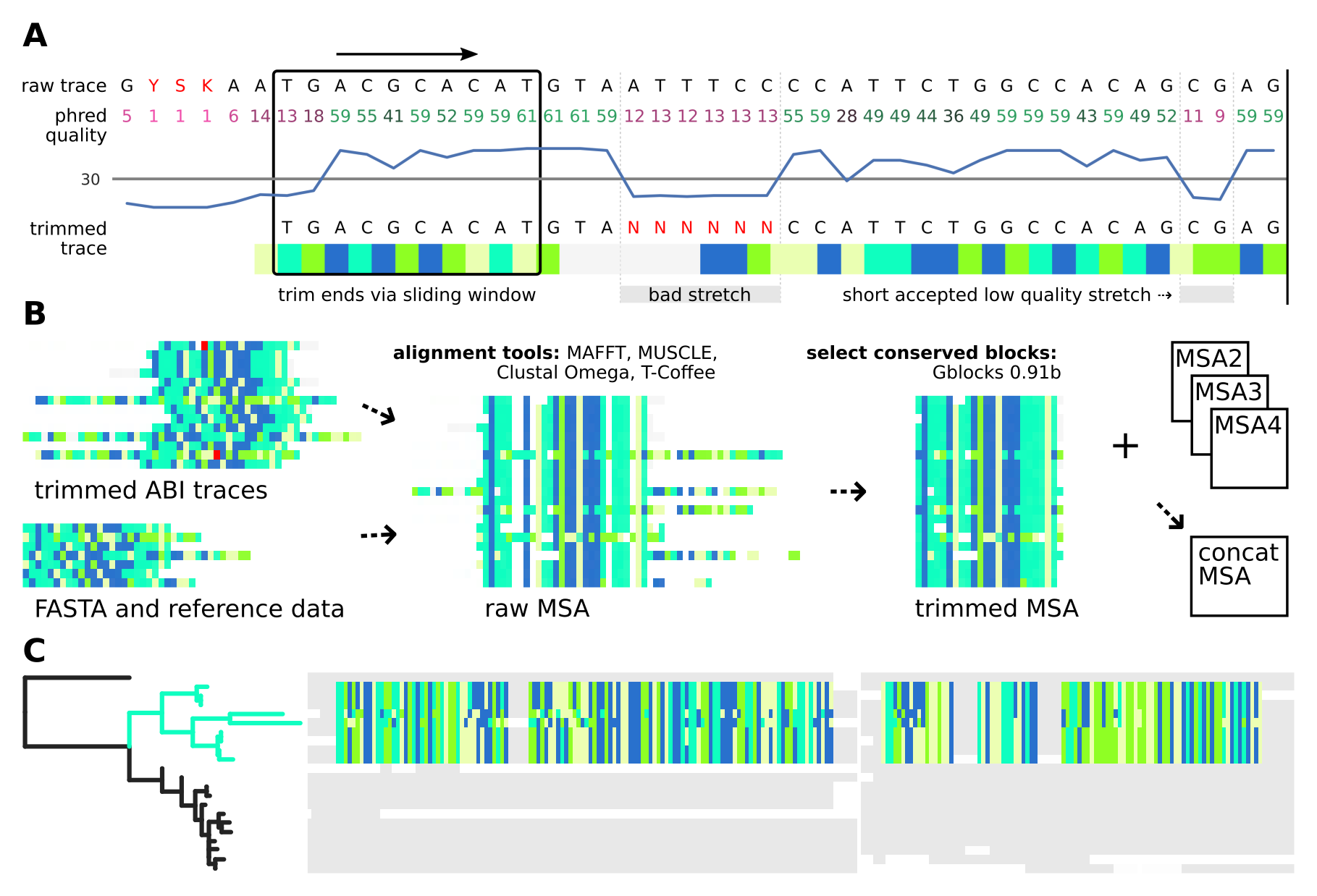
A:
Sequence data is extracted from ABI trace files using a customisable quality control: Sequence ends are trimmed with a sliding window until a certain number (8 out of 10 by default) of bases reach the minimal accepted phred quality score (between 0 and 60, 30 by default). Bases with low phred quality are replaced by N only if they form a consecutive stretch that is longer than a certain threshold (5 by default).
B:
Samples missing for a single locus are discarded for all genes. Trimmed traces as well as reference and FASTA sequences are aligned into single-gene Multiple Sequence Alignments (MSAs), which are then each trimmed to a user-defined level conserved positions using Gblocks 0.91b. For multi-gene analyses, the single-gene MSAs are then concatenated into a multi-gene MSA, which is used for ML tree inference. Trees are re-constructed using either RAxML-NG or IQ-Tree 2, with only the latter one available for Windows.
C:
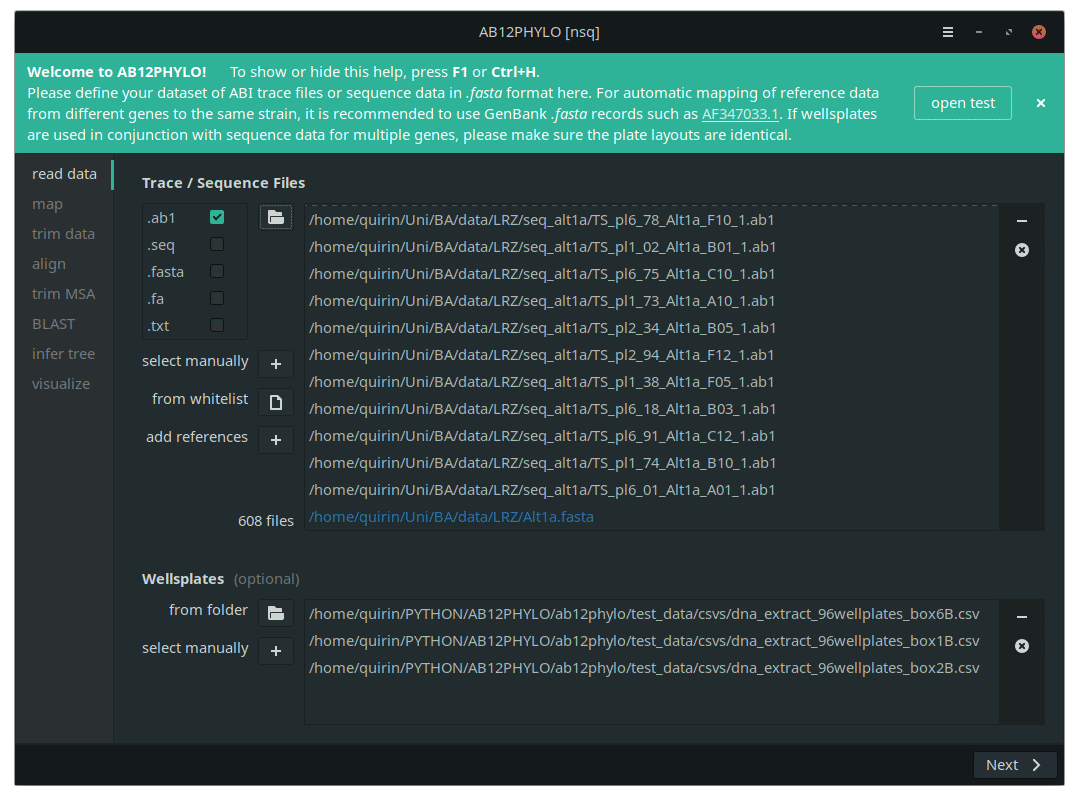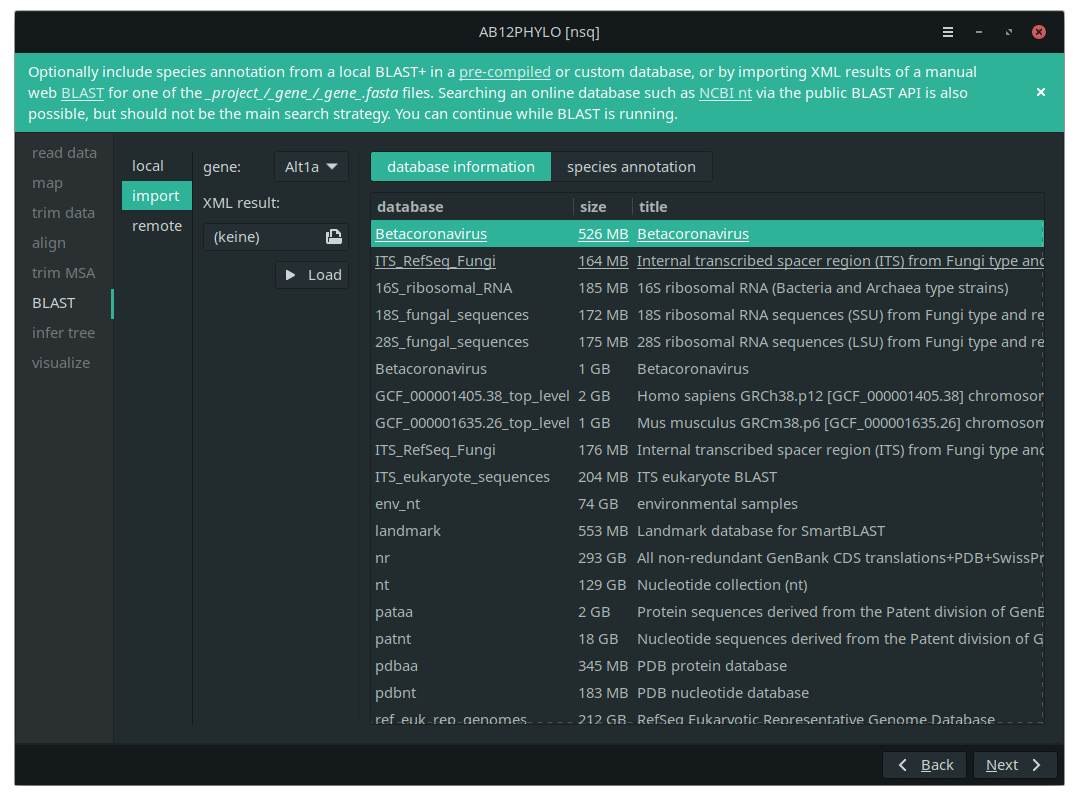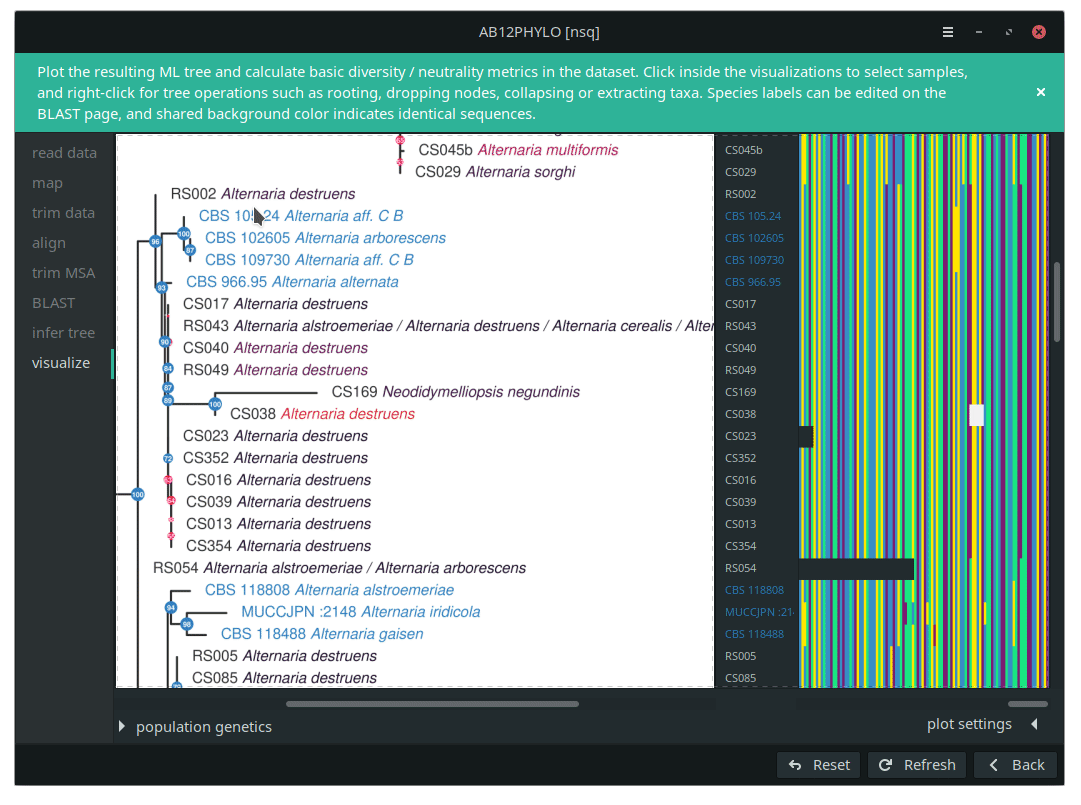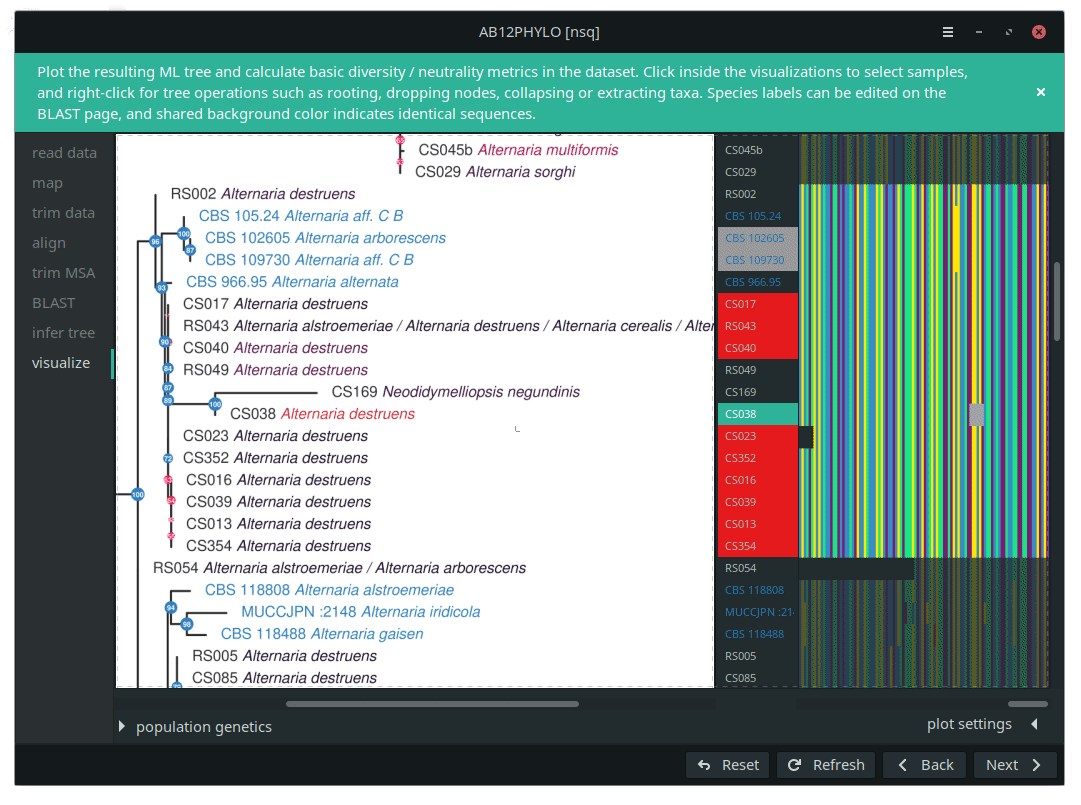
AB12PHYLO allows editing of the resulting tree and selection of taxa by label matching, shared ancestry or manual picking. For these selected sub-populations, basic population genetics neutrality and diversity metrics are calculated from the conserved MSA positions only, with adjustable tolerance of gaps and unknown characters. The graphical ab12phylo is both less cumbersome and more capable for these applications; the wiki pages (ab12phylo, ab12phylo-cmd) have more details.
A BLAST search for species annotation can be run on a local database, or via the public NCBI BLAST API. However, importing XML results of a web BLAST should be preferred to running remote API calls as a main strategy.
A simple ab12phylo-cmd example
A simple real-world invocation of commandline AB12PHYLO might look like this:
ab12phylo-cmd -abi <seq_dir> \
-csv <wellsplates_dir> \
-g <barcode_gene> \
-rf <ref.fasta> \
-bst 1000 \
-dir <results>
where:
<seq_dir>contains all input ABI trace files, ending in.ab1<wellsplates_dir>contains the.csvmappings of user-defined IDs to sequencer's isolate coordinates<barcode_gene>was sequenced, see here for more info<ref.fasta>contains full GenBank reference records like this- 1000
-bst=--bootstraptrees will be generated <results>is where results will be
Dependencies
Biopython, NumPy, pandas, Toytree <= 1.2.0, Toyplot, matplotlib, PyYAML, lxml, xmltramp2, svgutils, Pillow, Requests, Beautiful Soup and Jinja2
Non-python dependencies
The pipeline will use existing installations of the programs listed below if they are found on the system $PATH and not considered outdated. Otherwise, both ab12phylo and ab12phylo-cmd can download the latest static binaries from GitHub or the NCBI on their initial runs or if run with --initialize.
-
RAxML-NG version >=1.0.2
-
BLAST+ version >=2.9
-
an MSA tool: MAFFT, Clustal Omega, MUSCLE or T-Coffee
(clients for an EMBL service included) -
Gblocks 0.91b for MSA trimming (included)
References
-
Alexey M. Kozlov, Diego Darriba, Tomáš Flouri, Benoit Morel, and Alexandros Stamatakis (2019) RAxML-NG: A fast, scalable, and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics, btz305 doi:10.1093/bioinformatics/btz305
-
Nguyen,L. T., Schmidt,H. A., Von Haeseler,A., and Minh,B. Q. (2015) IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molecular Biology and Evolution, 32, 268–274. doi:10.1093/molbev/msu300
Release history Release notifications | RSS feed


Download files
Download the file for your platform. If you're not sure which to choose, learn more about installing packages.
Source Distribution
Built Distribution
Filter files by name, interpreter, ABI, and platform.
If you're not sure about the file name format, learn more about wheel file names.
Copy a direct link to the current filters
File details
Details for the file ab12phylo-0.5.21b0.tar.gz.
File metadata
- Download URL: ab12phylo-0.5.21b0.tar.gz
- Upload date:
- Size: 852.0 kB
- Tags: Source
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.4.2 importlib_metadata/4.6.1 pkginfo/1.7.0 requests/2.26.0 requests-toolbelt/0.9.1 tqdm/4.56.2 CPython/3.9.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
332772e9f5367e3aee4dc06a39f005a3075884214ab14a4c91044d59a5a99f75
|
|
| MD5 |
9505c80c3f58268913a1acca6bc57d51
|
|
| BLAKE2b-256 |
75c03049e38aebc0ac3baa275c336ad034c9fa08c501db8c39702054d3a641bd
|
File details
Details for the file ab12phylo-0.5.21b0-py3-none-any.whl.
File metadata
- Download URL: ab12phylo-0.5.21b0-py3-none-any.whl
- Upload date:
- Size: 986.1 kB
- Tags: Python 3
- Uploaded using Trusted Publishing? No
- Uploaded via: twine/3.4.2 importlib_metadata/4.6.1 pkginfo/1.7.0 requests/2.26.0 requests-toolbelt/0.9.1 tqdm/4.56.2 CPython/3.9.6
File hashes
| Algorithm | Hash digest | |
|---|---|---|
| SHA256 |
3efcc3efcc7c04b53642ed1e2f03d5e0871892eb1022ec2974dadadc04d259c1
|
|
| MD5 |
3d3813734a0cb653b30e09c94b5d93ad
|
|
| BLAKE2b-256 |
8f41251e28d976bb37c85d858164f25f60793c32c6b8187e9fb4e7f6adfa858a
|







